HRCT - Common diagnoses
Robin Smithuis, Otto van Delden and Cornelia Schaefer-Prokop
Radiology Department of the Rijnland Hospital, Leiderdorp and the Academical Medical Centre, Amsterdam, the Netherlands
Publicationdate
In this review we present the key findings in the most common interstitial lung diseases.
There are numerous interstitial lung diseases, but in clinical practice only about ten diseases account for approximately 90% of cases.
Knowledge of both radiological and clinical appearances of these more common interstitial lung diseases is therefore important for recognizing them in daily practice and including them in the differential diagnosis.
Some less common interstitial lung diseases will also be presented because their HRCT presentation may be very typical, allowing for a 'spot diagnosis' in selected cases.
In 'HRCT - basic interpretation' the terminology is introduced and a practical approach is given for the interpretation of HRCT examinations.
Introduction
More than 100 entities manifest as diffuse lung disease.
Fortunately only about 10 of these account for about 90% of all diffuse lung diseases, that are assessed by open lung biopsy.
Knowing the common and also uncommon HRCT-presentations of these frequently encountered diffuse lung diseases is extremely important.
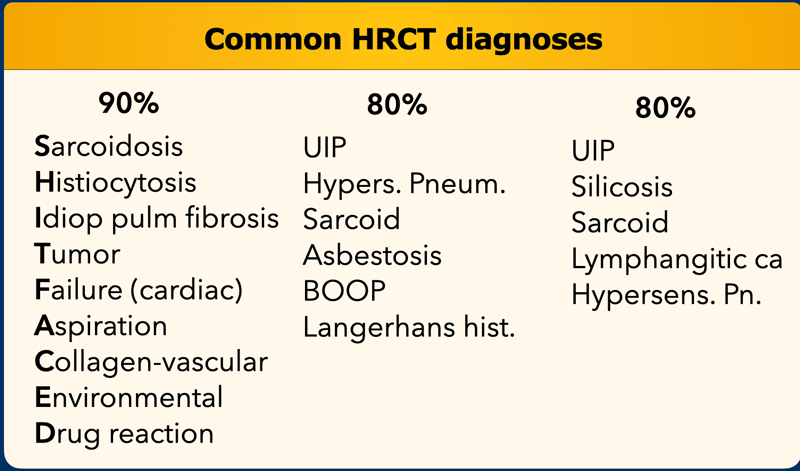
On the left you find three different lists of diagnoses.
Accounting for 80 - 90% of all diagnoses according to various literature references.
In some of them the old names are used and in some the newer ones.
The 'mnemonic' for the first list is 'SHIT FACED' (alternative shaded fit).
Sarcoidosis

Sarcoidosis is a systemic disorder of unknown origin.
It is characterized by non-caseating granulomas in multiple organs, that may resolve spontaneously or progress to fibrosis.
Pulmonary manifestations are present in 90% of patients.
Systemic symptoms such as fatigue, night sweats and weight loss are common.
Loefgren's syndrome, an acute presentation of sarcoidosis, consists of arthritis, erythema nodosum, bilateral hilar adenopathy and occurs in 9-34% of patients.
Erythema nodosum is seen predominantly in women and arthritis is more common in men.
Two third of patients have a remission within ten years.
One third have continuing disease leading to clinically significant organ impairment.
Less than 5% of patients die from sarcoidosis usually as a result of pulmonary fibrosis.
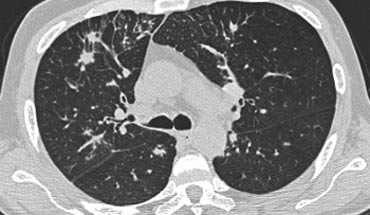
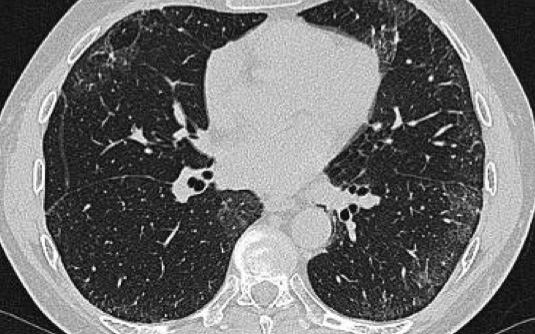
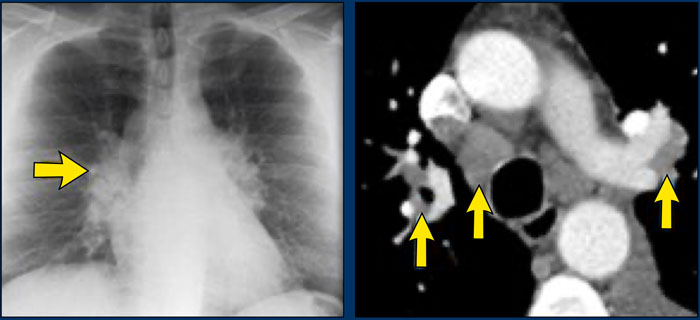 Sarcoidosis stage I: left and right hilar and paratracheal adenopathy (1-2-3 sign)
Sarcoidosis stage I: left and right hilar and paratracheal adenopathy (1-2-3 sign)
Stages
Chest films in sarcoidosis have been classified into four stages:
- Bilateral hilar lymphadenopathy
- Bilateral hilar lymphadenopath + pulmonary disease
- Only pulmonary disease
- Irreversible fibrosis
These stages do not indicate disease chronicity or correlate with changes in pulmonary function.
Images
A patient with stage I disease.
There is hilar and paratracheal adenopathy and no sign of pulmonary involvement.
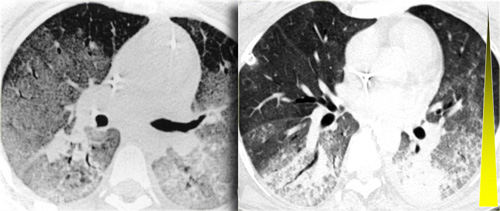
 Sarcoidosis: typical presentation with nodules along the bronchovascular bundle and fthe issuresNotice the partially calcified node in the left hilum.
Sarcoidosis: typical presentation with nodules along the bronchovascular bundle and fthe issuresNotice the partially calcified node in the left hilum.
HRCT findings in Sarcoidosis
- Common findings:
- Small nodules in a perilymphatic distribution (i.e. along subpleural surface and fissures, along interlobular septa and the peribronchovascular bundle).
- Upper and middle zone predominance.
- Lymphadenopathy in left hilus, right hilus and paratracheal (1-2-3 sign). Often with calcifications.
- Uncommon findings:
- Conglomerate masses in a perihilar location.
- Larger nodules (> 1cm in diameter, in Grouped nodules or coalescent nodlues surrounded by multiple satellite nodules (Galaxy sign)
- Nodules so small and dense that they appear as ground glass or even as consolidations (alveolar sarcoidosis)
Image
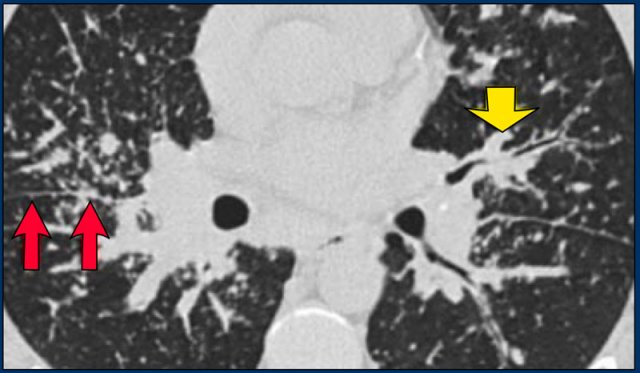
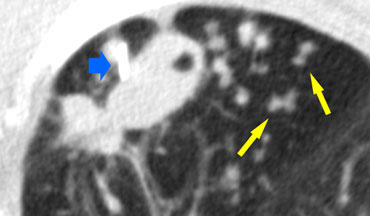
A typical presentation of sarcoidosis with hilar lymphadenopathy and small nodules along bronchovascular bundles (yellow arrow) and along fissures (red arrows).

Image
A detailed view with the typical HRCT-presentation with nodules along bronchovascular bundle (red arrow) and fissures (yellow arrow).
This is the typical perilymphatic distribution of the nodules.
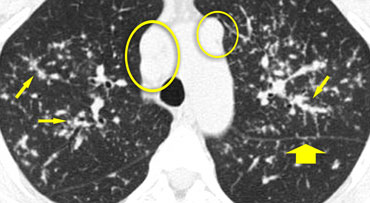 Sarcoidosis: typical presentation
Sarcoidosis: typical presentation
The HRCT appearance of pulmonary sarcoidosis varies greatly and is known to mimic many other diffuse infiltrative lung diseases.
Approximately 60 to 70% of patients with sarcoidosis have characteristic radiologic findings.
In 25 to 30% of cases the radiologic findings are atypical.
In 5 to 10% of patients the chest radiograph is normal.
On the left another typical presentation of sarcoidosis with mediastinal lymphadenopathy and small nodules in a perilymphatic distribution along bronchovascular bundles and along fissures (yellow arrows).
Always look for small nodules along the fissures, because this is a very specific and typical sign of sarcoidosis.
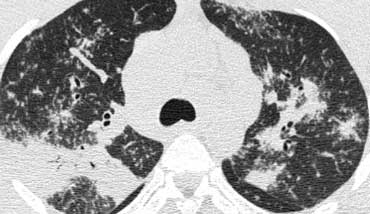 Sarcoidosis with conglomerate masses of fibrous tissue
Sarcoidosis with conglomerate masses of fibrous tissue
Fibrosis in Sarcoidosis.
Progressive fibrosis in sarcoidosis may lead to peribronchovascular (perihilar) conglomerate masses of fibrous tissue.
The typical location is posteriorly in the upper lobes, leading to volume loss of the upper lobes with displacement of the interlobar fissure.
Other diseases that commonly result in this appearance are:
- Silicosis
- Tuberculosis
- Talcosis
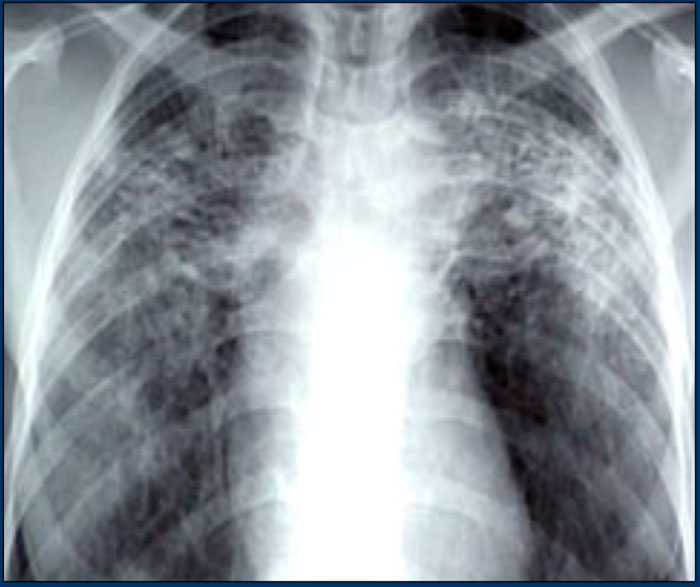 Sarcoidosis with fibrosis in the upper lobes. Typical chest film.
Sarcoidosis with fibrosis in the upper lobes. Typical chest film.
Here a typical chest film of long standing sarcoidosis (stage IV) with fibrosis in the upper zones and volume loss of the upper lobes resulting in hilar elevation.
Fibrosis results in obliteration of pulmonary vessels, which can lead to pulmonary hypertension.
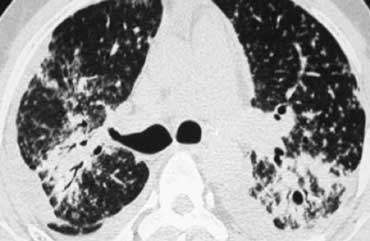 Sarcoidosis with fibrosis in the upper lobes. Typical HRCT findings.
Sarcoidosis with fibrosis in the upper lobes. Typical HRCT findings.
Here another case of stage IV sarcoidosis.
Notice the distribution of the conglomerate masses of fibrosis in the posterior part of the lungs.
In addition there are multiple small well-defined nodules.
Some of these nodules have the typical subpleural distribution.
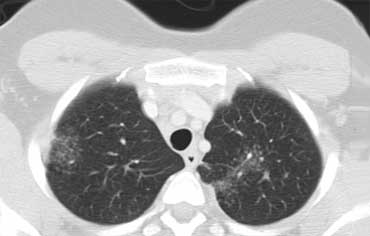
Alveolar Sarcoidosis.
This is a case of alveolar sarcoidosis.
Scroll through the images.
The appearance resembles a ground glass attenuation, but with a closer look you may appreciate that the increased attenuation is the result of many tiny grouped nodules.
Also notice the hilar lymphadenopathy.
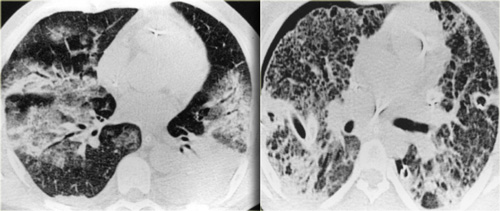
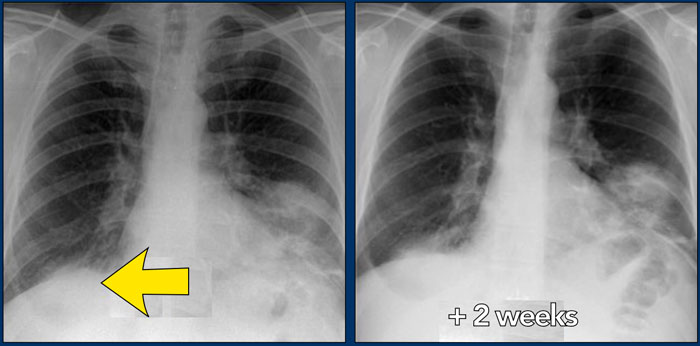 47-year old female patient treated for possible infection
47-year old female patient treated for possible infection
Alveolar Sarcoidosis (2)
On the left a 47-year old female patient with a dry cough, slightly breathless and a normal blood analysis.
A chest film was taken and she was treated with antibiotics.
A follow up film was made, because she did not improve.
The first chest film shows bilateral consolidations in the lower lobes (arrow), initially interpreted as infection.
After two weeks of treatment with antibiotics, there is no improvement.
The differential diagnosis now includes tumor (bronchoalveolar carcinoma or lymphoma), eosinophilic pneumonia , organizing pneumonia, Wegener's disease or an uncommon presentation of sarcoidosis.
Now continue with the HRCT.
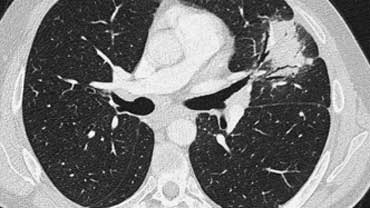
Scroll through the images on the left .
There are multiple areas of consolidation.
Ancillary findings are hilar and mediastinal lymphadenopathy.
The differental diagnosis of the CT-images is basically the same as of the chest film.
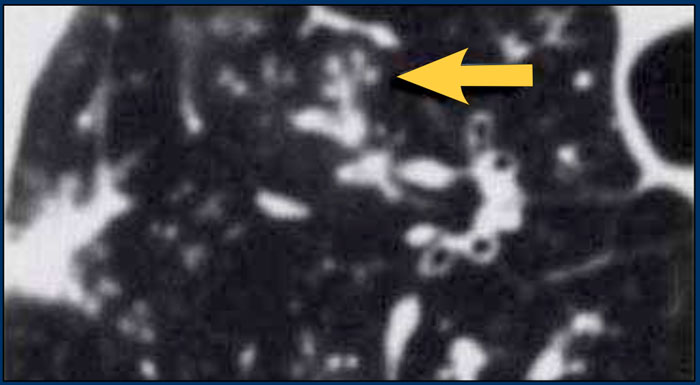
Histology revealed alveolar sarcoid.
There is only one clue to the diagnosis and that is the presence of small nodules that can be identified in image 3, but these are difficult to see.
This case nicely demonstrates that sarcoidosis truely is 'the great mimicker'.
Sarcoidosis should be therefore in our differential diagnostic list!.
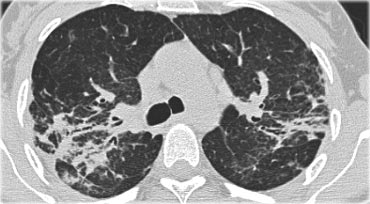
On the left a case of fibrosing sarcoidosis, showing fibrosis, traction bronchiectases and crowding of the involved bronchi, predominantly in the perihilar region and upper lobes.
Nodular abnormalities are absent, but the appearance and the location of the fibrosis are very suggestive of the diagnosis of sarcoidosis.
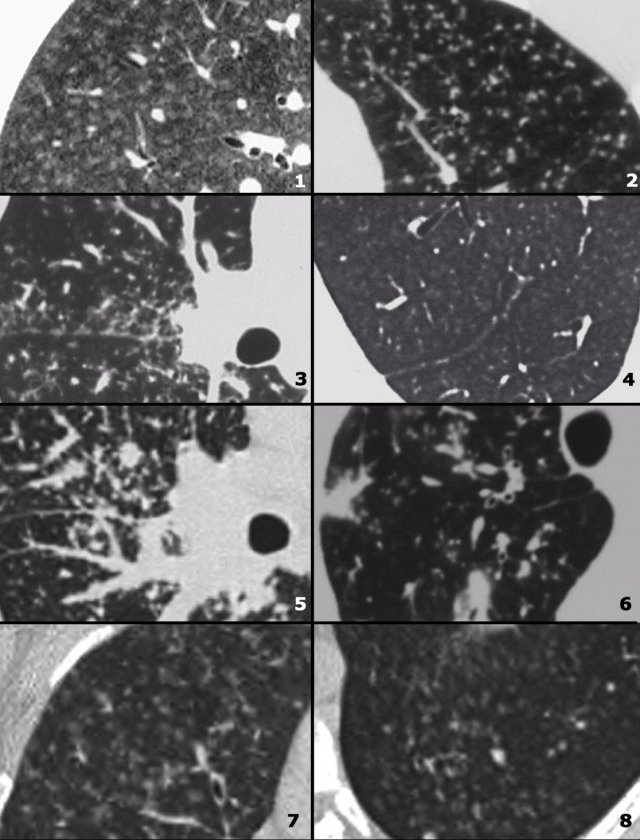
Differential diagnosis of Sarcoidosis.
- Lymphadenopathy:
- Primary TB: asymmetrical adenopathy
- Histoplasmosis
- Lymphoma
- Small cell lung cancer with nodal metastases
- Nodular pattern:
- Silicosis / Pneumoconiosis: predominantly centrilobular and subpleural nodules.
- Miliary TB: random nodules.
- Fibrotic pattern:
- Usual Interstitial Pneumonia (UIP): basal and peripheral fibrosis, honeycombing.
- Chronic Hypersensitivity Pneumonitis: mid zone fibrosis with mosaic pattern.
- Tuberculosis (more unilateral).
On the left some diseases with a nodular pattern.
- Hypersensitivity pneumonitis: ill defined centrilobular nodules.
- Miliary TB: random nodules of the same size.
- Sarcoidosis: nodules with perilymphatic distribution, along fissures, adenopathy.
- Hypersensitivity pneumonitis: centrilobular nodules, notice sparing of the subpleural area.
- Sarcoidosis: nodules with perilymphatic distribution, along fissures, adenopathy.
- TB: Tree-in-bud appearance in a patient with active TB.
- Langerhans cell histiocytosis: early nodular stage before the typical cysts appear.
- Respiratory bronchiolitis: ill defined centrilobular nodules of ground-glass opacity.
Silicosis / Coal worker pneumoconiosis
Silicosis and Coal worker pneumoconiosis (CWP) are pathologically distinct entities with differing histology, resulting from the inhalation of different inorganic dusts.
The radiographic and HRCT appearances of these diseases, however, may not be distinguishable from each other and may be similar to sarcoidosis.
It is important to realize that these diseases are rare compared to sarcoidosis.
Silicosis and CWP occur in a specific patient group (construction workers, mining workers, workers exposed to sandblasting, glass blowing and pottery).
HRCT findings in Silicosis/CWP
- Small well-defined nodules of 2 to 5mm in diameter in both lungs.
- Upper lobe predominance
- Nodules may be calcified
- Centrilobular and subpleural distribution
- Sometimes random distribution
- Irregular conglomerate masses, known as progressive massive fibrosis
- Masses may cavitate due to ischemic necrosis.
- Often hilar and mediastinal lymphnodes.
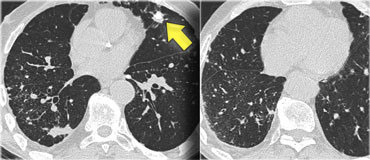
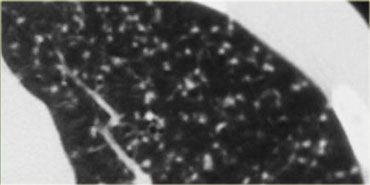
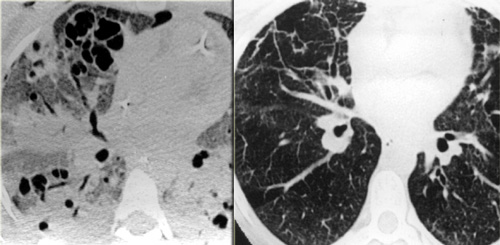
On the left a case of silicosis showing nodules of varying sizes with a random and subpleural distribution. One nodule contains calcification (arrow). Note the absence of a lymphatic distribution pattern (peribronchovascular and along fissures), which would be suggestive of sarcoidosis.
 Same patient with silicosis as previous images showing a conglomerate mass in a perihilar location in the right upper lobe. The left lobe shows multiple nodules of varying size.
Same patient with silicosis as previous images showing a conglomerate mass in a perihilar location in the right upper lobe. The left lobe shows multiple nodules of varying size.
Differential diagnosis of Silicosis / Pneumoconiosis.
- Sarcoidosis : can be difficult to distinguish (look for distribution of nodules).
- Infection: miliairy TB, fungus.
- Hematogenous metastases: silicotic nodules in subpleural and peribronchiolar location up to the level of the secundary pulmonary lobule, may have a seemingly random distribution and simulate metastases and miliary infections.
- Langerhans cell histiocytosis: can be difficult to distinguish from silicosis in the early stage, when LCH is solely characterized by the presence of small nodules. Look for nodules with cavitation.
Lymphangitic Carcinomatosis

Lymphangitic Carcinomatosis results from hematogenous spread to the lung, with subsequent invasion of interstitium and lymphatics.
The presenting symptoms are dyspnea and cough and can predate the radiographic abnormalities.
In many cases however the patients are asymptomatic.
Lymphangitic Carcinomatosis is seen in carcinoma of the lung, breast, stomach, pancreas, prostate, cervix, thyroid and metastatic adenocarcinoma from an unknown primary.
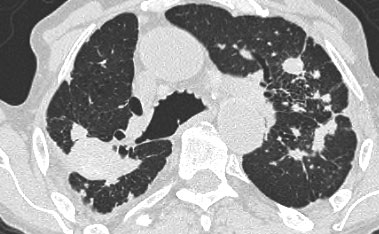
HRCT findings in Lymphangitic Carcinomatosis
- Interlobular septal thickening, thickening of fissures and thickening of the peribronchovascular interstitium (bronchial cuffing).
- Depending on filling with fluid or with tumor cells, septal thickening is irregular or smooth.
- Focal or unilateral abnormalities in 50% of patients.
- Hilar lymphadenopathy in 50% of patients.
- Pleural effusion due to pleuritic carcinomatosis ( > 50% of patients).
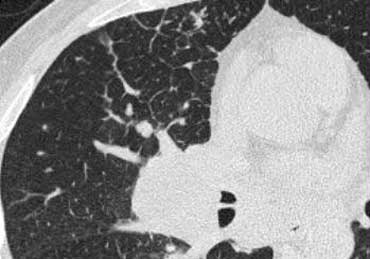
Image
A patient with Lymphangitic Carcinomatosis.
Notice the focal distribution.
This finding is helpful in distinguishing Lymphangitic Carcinomatosis from other causes of interlobular septal thickening like pulmonary edema or sarcoid.
There is also lymphadenopathy.
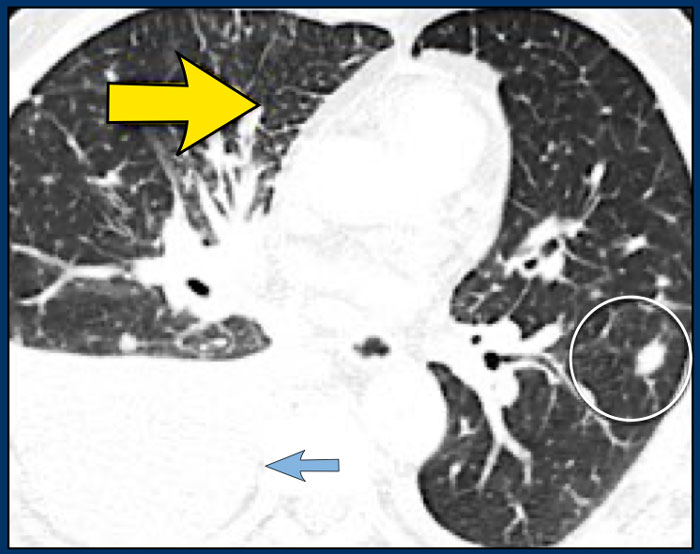
Image
Another patient with Lymphangitic Carcinomatosis with interlobular septal thickening (yellow arrow).
Additional pleural fluid (blue arrow) and a lung metastasis (circle).
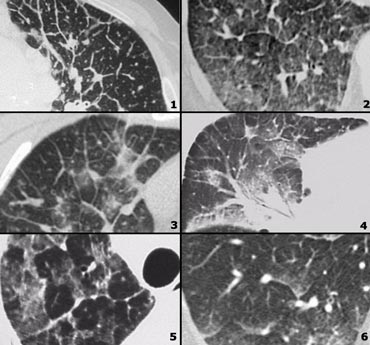
Examples of Lymphangitic carcinomatosis and differential diagnosis
- Lymphangitic carcinomatosis: irregular septal thickening, usually focal or unilateral, in 50% adenopathy, known carcinoma.
- Cardiogenic pulmonary edema: bilateral abnormalities, filling of alveoli, enlarged heart, rapid response to diuretics, ground-glass opacity due to filling of alveoli with fluid, gravitational distribution of the alveolar fluid.
- Lymphangitic carcinomatosis.
- Lymphangitic carcinomatosis with hilar adenopathy and thickening of the central bronchovascular interstitium.
- Alveolar proteinosis: sharply demarcated secondary lobeles with ground glass attenuation as opposed to secondary lobules with normal aeration, superimposed inter and intralobular septal thickening (crazy paving).
- Cardiogenic pulmonary edema.
Cardiogenic pulmonary edema

Patients with pulmonary edema are not imaged with HRCT as their diagnosis is usually based on a combination of clinical and chest radiographic findings.
However sometimes the diagnosis is not that straightforward and knowledge of the HRCT appearance of pulmonary edema can be helpful in avoiding misdiagnosis.
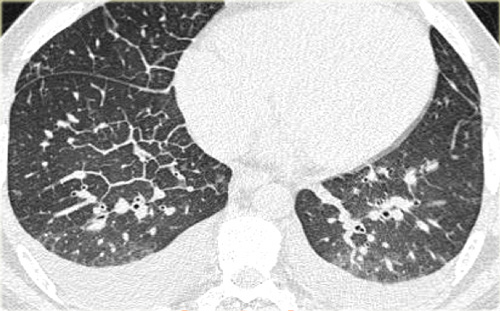 Cardiogenic pulmonary edema
Cardiogenic pulmonary edema
HRCT findings in cardiogenic pulmonary edema
- Bilateral septal thickening and ground-glass opacity.
- Perihilar and gravitational distribution predominatly in the dependent lung.
- Cardiomegaly and pleural fluid.
On the left typical features of cardiogenic pulmonary edema
There is smooth septal thickening and some ground glass opacity in the dependent part of the lungs.
In addition there is bilateral pleural fluid.
In a patient with a known malignancy lymphangitic carcinomatosis would be high in the differential diagnostic list.
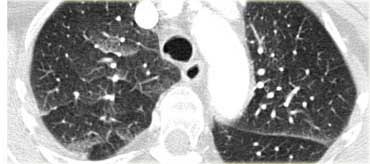 Cardiogenic pulmonary edema
Cardiogenic pulmonary edema
Differential diagnosis of cardiogenic pulmonary edema.
- Lymphangitic carcinomatosis
- Interstitial pneumonia (viral, mycoplasma)
- ARDS
- Pulmonary hemorrhage
On the left another example of cardiogenic pulmonary edema.
This patient had a CT to rule out pulmonary emboli.
There is smooth septal thickening and ground glass opacity in a more patchy distribution.
Note: edema can have a very unusual appearance and be distributed very patchy: some areas are filled with fluid as opposed to other areas in immediate vicinity which appear normal.
Hypersensitivity Pneumonitis

Hypersensitivity pneumonitis (HP) is also known as extrinsic allergic alveolitis (EAA).
HP is an allergic lung disease caused by the inhalation of a variety of antigens (farmer's lung, bird fancier's lung, 'hot tub' lung, humidifier lung).
The radiographic and pathologic abnormalities in patients can be classified into acute, subacute, and chronic stages.
Mostly HRCT is performed in the subacute stage of HP, weeks to months following the first exposure to the antigen or in the chronic phase.
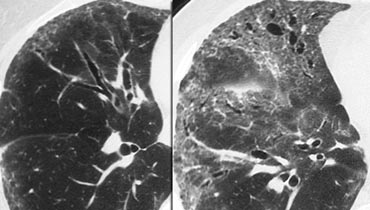
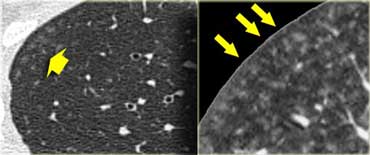 Subacute hypersensitivity pneumonitis with ill-defined centrilobular nodules
Subacute hypersensitivity pneumonitis with ill-defined centrilobular nodules
Subacute hypersensitivity pneumonitis
The key findings in the subacute hypersensitivity pneumonitis are:
- ill-defined centrilobular nodules of ground-glass opacity (80% of cases) or
- mosaic pattern of a combination of patchy ground-glass opacity due to lung infiltration and patchy lucency due to bronchiolitis with air trapping
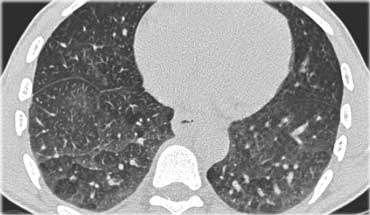
Here two examples of subacute hypersensitivity pneumonitis.
There are ill-defined centrilobular nodules of ground-glass opacity.
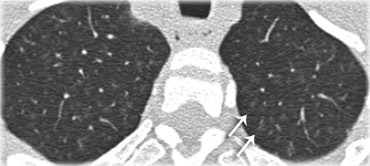
Here another case of subacute hypersensitivity pneumonitis.
There is subtle opacity in the centre of the secondary lobules (arrows) with sparing of the subpleural region.
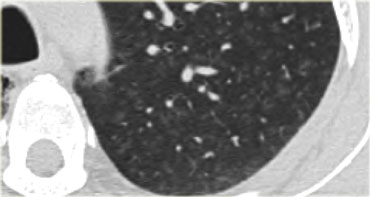
This HRCT-image also demonstrates subtle centrilobular opacity in a patient with subacute HP.
Notice how ill-defined these centrilobular nodules are.
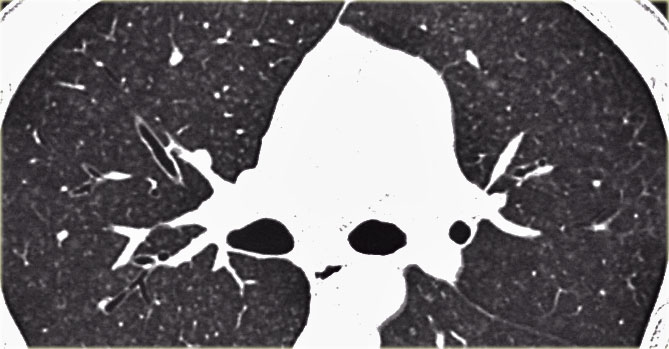
Sometimes the centrilobular opacities are more nodular in appearance as in this case.
 Subacute hypersensitivity pneumonitis with mosaic pattern
Subacute hypersensitivity pneumonitis with mosaic pattern
Here another case of hypersensitivity pneumonitis.
There is a mosaic pattern.
Some secundary lobules demonstrate ground-glass opacity due to lung infiltration, while others are more lucent due to bronchiolitis with air trapping.
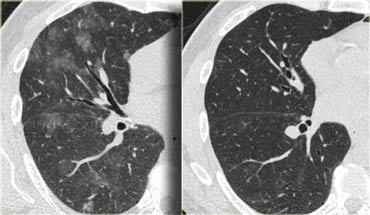 LEFT: HRCT at presentation. RIGHT: HRCT ten days later
LEFT: HRCT at presentation. RIGHT: HRCT ten days later
This patient presented with acute dyspnoe and a normal chest film (not shown).
The HRCT at presentation (left) shows lobular areas of ground glass attenuation.
A control HRCT ten days later (right) demonstrated, that the findings had resolved without any treatment.
The findings were thought to be due to hypersensitivity pneumonitis.
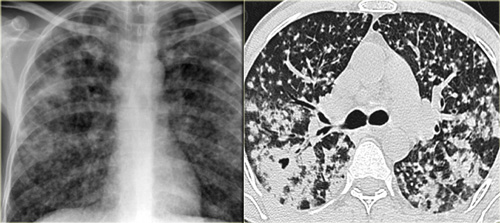
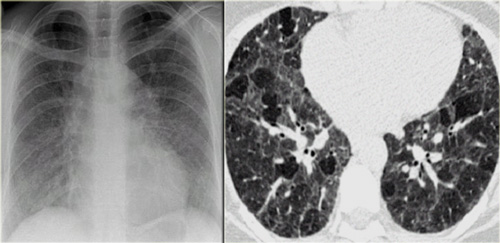 Chronic hypersensitivity pneumonitis: Nonspecific chest film, typical HRCT-findings.
Chronic hypersensitivity pneumonitis: Nonspecific chest film, typical HRCT-findings.
Chronic hypersensitivity pneumonitis
The key findings in chronic hypersensitivity pneumonitis are:
- Mosaic pattern with areas of ground-glass atenuation and areas of low attenuation.
- Fibrosis and parenchymal distortion in a mid zone distribution.
On the left a patient with chronic hypersensitivity pneumonitis.
The HRCT shows a mosaic pattern with hyperaerated secondary nodules and secondry nodules of increased attenuation.
Additionally there is septal and intralobular reticular thickening, indicating already existing irreversible fibrosis.
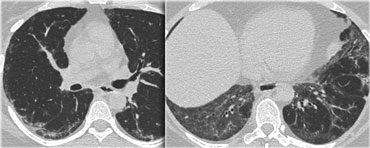
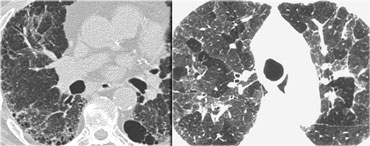 UIP with honeycombing (left) and chronic HP (right)
UIP with honeycombing (left) and chronic HP (right)
Differential diagnosis of Hypersensitivity Pneumonitis.
-
Subacute stage:
- RB-ILD: seen in smokers, upper lobe predilection, usually associated with centrilobular emphysema.
- Alveolar proteinosis.
-
Chronic stage:
-
UIP: may show very similar HRCT findings.
UIP has a strong lower zone distribution.
In chronic HP fibrotic changes are typically seen throughout the whole lung parenchyma from the periphery towards the centrum.
-
UIP: may show very similar HRCT findings.
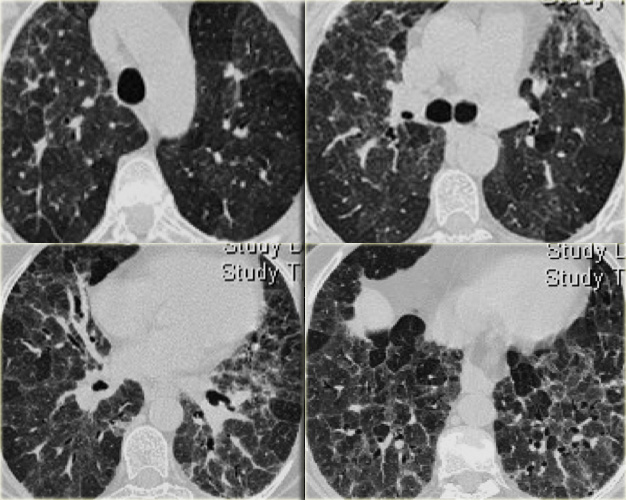 Chronic hypersensitivity pneumonitis.
Chronic hypersensitivity pneumonitis.
Chronic hypersensitivity pneumonitis (2)
The case on the left shows an inspiratory and expiratory scan: the mosaic pattern with areas of ground-glass attenuation and areas of low attenuation, that become more evident on the expiratory scan, indicating air trapping.
Signs of fibrosis such as distorted vessels and bronchi as well as septal thickening are more pronounced in the mid and lower lung zones, but not limited to the subpleural area.
The images on the left suggest the diagnosis hypersensitivity pneumonitis.
Based on the imaging findigs alone, alveolar proteinosis and other diseases with a mozaic pattern should be included in the differential diagnosis.
Tuberculosis
Primary TB:
Initial infection with consolidation, adenopathy and pleural effusion.
Secondary TB :
Post-primary or reactivation TB.
This is the reactivation of the original infection.
Usually located in the apical segments of upper lobes with cavitation
Endobronchial spread: May occur in both primary and secondary TB, when the infection is not contained.
Hematogenous spread (miliary TB): May occur in both primary and secondary TB, when the infection is not contained.
 TB: cavitating lesion and endobronchial spread
TB: cavitating lesion and endobronchial spread
HRCT findings in TB
-
Primary TB:
- Consolidation anywhere, lymphadenopathy and pleural effusion. Usually regresses to calcified lung nodule and calcified ipsilateral lymph node.
- Secondary TB:
- Consolidation in apical segments of upper lobes or superior segments of lower lobes.
- Cavitation
- Endobronchial spread:
- Tree in bud appearance
- Miliary TB:
- 2-3 mm nodules with random disrtibution.
On the left a patient with TB. There is a cavitating lesion and typical tree-in-bud appearance. The blue arrow indicates the biopsy needle.
 Miliary TB
Miliary TB
Miliary TB
This represents a hematogenous dissemination of infection and may occur in association with either primary or postprimary disease.
It is characterized by uniform small nodules with a random distribution.
 TB with cavitation
TB with cavitation
Cavitation in TB
The chest film on the left shows diffuse areas with nodular air space opacifications.
The HRCT demonstrates multiple nodules in peribronchial distribution, partially confluent, and a cavitation in the right lung, strongly suggestive for tuberculosis.
Other diseases in the differential are Wegener granulomatosis or malignancy (both show no tree-in-bud).
 Endobronchial spread of TB with tree-in-bud
Endobronchial spread of TB with tree-in-bud
Endobronchial spread of TB
This can occur with primary or postprimary infection.
In most subjects, the primary infection is localized and clinically inapparent.
However, in 5 to 10% of patients with primary TB, the infection is poorly contained and dissemination occurs.
This is termed progressive primary tuberculosis.
Extensive cavitation of the tuberculous pneumonia lead to endobronchial spread of the infection.
Rupture of necrotic lymph nodes into the bronchi can also result in endobronchial dissemination.
Tree-in-bud appearance is typical for active endobronchial spread of infection.
It occurs in acute tuberculosis but also in any other bacterial infection.
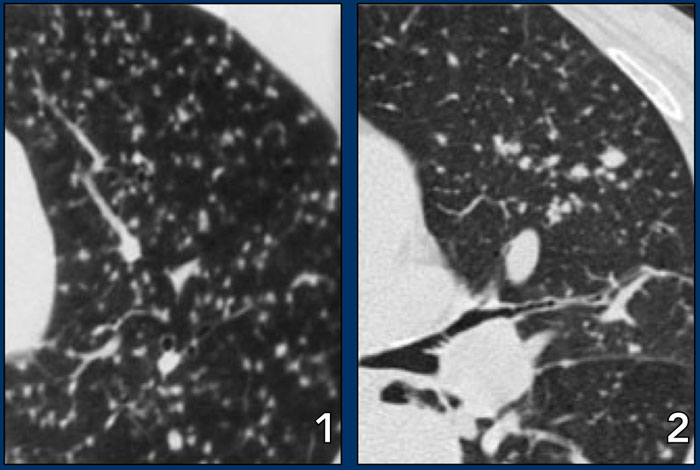 LEFT: miliary TB. RIGHT: metastases
LEFT: miliary TB. RIGHT: metastases
Differential diagnosis of TB.
- Primary TB: Acute bacterial pneumonia
- Secondary TB: Sarcoidosis, Silicosis, Pneumoconiosis
- Endobronchial spread of TB: Bronchopneumonia, Hypersensitivity pneumonitis
- Miliary TB: metastases of medullary thyroid ca, chorionca and melanoma.
In both miliary TB and metastases the nodules have a random distribution.
In miliary TB the nodules are more uniform in size.
Images
- TB - uniform small nodules in random distribution
- Metastases - nodules of different sizes in random distribution.
Chronic eosinophilic pneumonia

Chronic eosinophilic pneumonia is an idiopathic condition characterized by filling of the alveoli with eosinophils.
It is associated with an increased number of eosinophils in the peripheral blood and patients present with fever, cough, weight loss, malaise, and shortness of breath.
The symptoms are often severe and last three months or more.
Patients respond promptly to treatment with steroids.
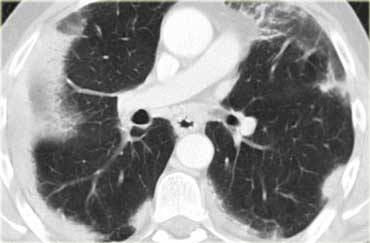 Chronic eosinophilic pneumonia
Chronic eosinophilic pneumonia
HRCT findings in Chronic eosinophilic pneumonia
- Peripheral consolidations with upper lobe predominance (photo negative of pulmonary edema).
On the left a contrast enhanced CT in a patient with chronic eosinophilic pneumonia.
Notice peripheral distribution of the consolidations.
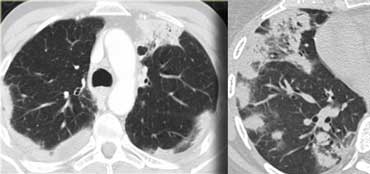 Chronic eosinophilic pneumonia (left) versus Organizing pneumonia (right)
Chronic eosinophilic pneumonia (left) versus Organizing pneumonia (right)
Differential diagnosis of Chronic eosinophilic pneumonia
- Organizing pneumonia (COP)
- Loffler syndrome (eosinophilia and vanishing peripheral consolidations)
- Churg-Strauss syndrome (also serum eosinophilia, asthma, systemic vasculitis affecting multiple organs: renal insufficiency, arthralgia and myocarditis and pericarditis)
- Pulmonary infarcts
The images on the left show the similarities between chronic eosinophilic pneumonia and organizing pneumonia.
Differentiation has to be made on the basis of clinical and laboratory findings.
Pneumocystis carinii pneumonia
Pneumocystis carinii pneumonia (PCP) or pneumocystis jiroveci as it is currently named, is an opportumistic infection in immunocompromised patients.
PCP used to affect most HIV-infected patients at some point during the course of their disease, but with the new anti-viral drugs it has become less common.
Nowadays PCP is seen more in immunosuppressed patients, i.e. transplant recipients and patients on chemotherapy.
HRCT findings in PCP
- Perihilar or diffuse ground-glass opacification.
- Sometimes thickened septal lines in association with areas of ground-glass
- Later cysts (or pneumatoceles) in 10-35% of patients, typically involving upper lobes
- Cysts may have bizarre shapes and thick walls
- Following therapy these lesions eventually regress, resulting either in complete disappearance, or residual nodules or scars
- Pneumothorax in 35% of patients with cysts
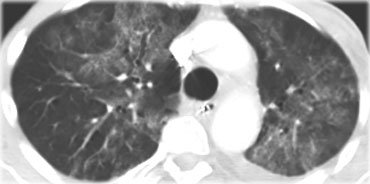 PCP with diffuse ground-glass opacification
PCP with diffuse ground-glass opacification
On the left an immunocompromised patient with PCP.
The CT findings are diffuse ground-glass opacification.
The findings are not specific for PCP, but in this clinical setting PCP is the most likely diagnosis.
On the left another patient with PCP.
Scroll through the images.
ARDS
Acute respiratory distress syndrome (ARDS) is a sudden, life-threatening lung failure requiring mechanical ventilation.
ARDS represents the result of increased permeability often in combination with injury to the respiratory epithelium.
A variety of underlying conditions, from infections to major trauma, can cause ARDS.
Primary pulmonary risk factors include aspiration, pneumonia, toxic inhalation and pulmonary contusion.
Extrapulmonary risk factors are sepsis, pancreatitis, multiple blood transfusions, trauma and the use of drugs such as heroin.
Mild forms of ARDS may resolve completely, while severe forms result in irreverible fibrosis.
Why some people develop ARDS and others do not is unknown.
 Extra-pulmonary ARDS with gravity dependent gradient
Extra-pulmonary ARDS with gravity dependent gradient
Extra-pulmonary ARDS
On the left a patient who was involved in a traffic accident and within hours developed ARDS.
The dominant pattern is ground glass opacity.
In the dependent parts of the lung there is also some consolidation, so there is a gradient from front to back.
An important finding in extra-pulmonary ARDS is the symmetry of the abnormalities.
 Pulmonary ARDS with asymmetric patchy distribution of consolidations.
Pulmonary ARDS with asymmetric patchy distribution of consolidations.
Pulmonary ARDS
On the left a patient who developed ARDS as a result of pneumonia (i.e. pulmonary ARDS).
Note the patchy distribution of lung disease and the almost complete distorsion more basal.
Patient is ventilated with PEEP (positive end expiratory pressure ) leading to a barotrauma of the lung parenchyma: there are multiple subpleural cysts and a bilateral pneumothorax.
 LEFT: ARDS. RIGHT: Fibrosis in the anterior parts as a result of damage related to barotraumas and PEEP ventilation
LEFT: ARDS. RIGHT: Fibrosis in the anterior parts as a result of damage related to barotraumas and PEEP ventilation
Consolidations have a protecting effect on the lung parenchyma under PEEP ventilation, while the ventrally located areas of more normal lung are most prone to the effects of barotrauma.
As a result we find cystic destruction ventrally and residual fibrosis mostly in the ventral lung areas.
Idiopathic interstitial pneumonias

The idiopathic interstitial pneumonias (IIPs) comprise a heterogenous group of disorders.
They represent fundamental responses of the lung to injury and do not represent 'diseases' per se.
Idiopathic indicates unknown cause and interstitial pneumonia refers to involvement of the lung parenchyma by varying combinations of fibrosis and inflammation.
IIPs include seven entities listed in the table on the left in order of relative frequency.
These diseases have specific patterns of morphologic findings on HRCT and histology.
Before we call these findings idiopathic or cryptogenic, we should realise, that these patterns are also common findings in collagen vascular diseases (e.g., sclerodermia, rheumatoid arthritis) and drug-related lung diseases.
For instance in patients with rheumatoid arthritis findings of NSIP, UIP, OP and LIP have been reported.
UIP
Usual Interstitial Pneumonitis (UIP) is a histologic diagnosis.
UIP has distinctive HRCT findings and is usually shown at lungbiopsy, when honeycombing is visible.
If the UIP pattern is of unknown cause (i.e. idiopathic), the disease is called Idiopathic pulmonary fibrosis (IPF).
IPF accounts for more than 60% of the cases of UIP.
In the presence of a surgical biopsy showing a UIP pattern the diagnosis of IPF requires exclusion of other known causes of UIP including drug toxicities, environmental exposures (asbest), and collagen vascular diseases like RA, SLE, polyarteritis nodosa and sclerodermia.
A long list of drugs have been implicated, but this pattern is most commonly the result of cytotoxic chemotherapeutic agents such as bleomycin, busulfan, vincristine, methotrexate, adriamycin, and carmustine (BCNU).
The differentiation between NSIP and UIP has tremendous prognostic implication for the patient.
UIP is more progressive and more than 50% of patients with UIP die within 3 years.
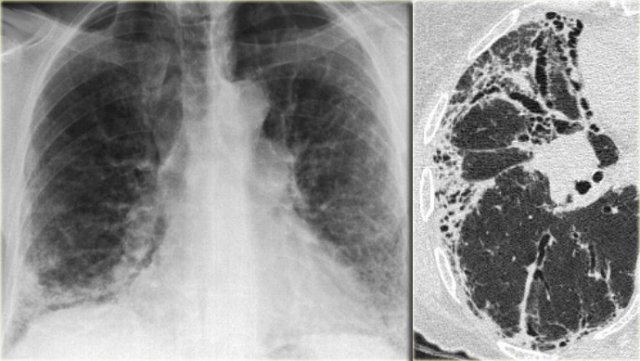 Chest film in a patient with UIP demonstrating the reticular pattern in basal and subpleural distribution due to honeycombing.
Chest film in a patient with UIP demonstrating the reticular pattern in basal and subpleural distribution due to honeycombing.
On the left a chest film of a patient with UIP due to IPF.
The findings on the chest film comprise volume loss and fibrotic changes in the basal lung area.
The radiographic appearance of honeycombing comprises reticular densities caused by the thick walls of the cysts.
Whenever you see a chest film with long standing reticulation with a lower lobe and peripheral preference also think 'UIP'.
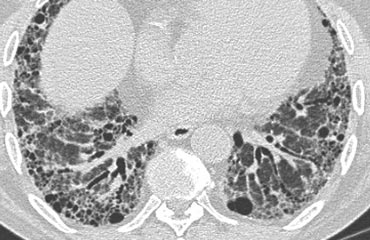 Typical UIP
Typical UIP
HRCT findings in UIP
- Honeycombing consisting of multilayered thick-walled cysts.
- Architectural distortion with traction bronchiectasis due to fibrosis.
- Predominance in basal and subpleural region.
- Mild mediastinal lymphadenopathy
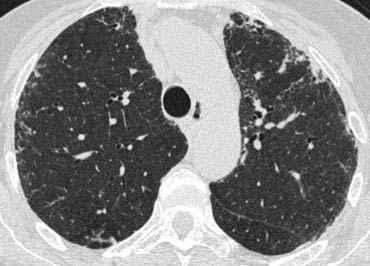
Differential diagnosis of UIP.
- Chronic Hypersensitivity Pneumonitis
- End stage Sarcoidosis
- NSIP
Chronic HP may be indistinguishable.
It is suspected if there is a mosaic pattern with sparing of the lung bases or when there are centrilobular nodules.
Sarcoidosis is a more likely diagnosis if the fibrosis is located in the posterior parts of the upper lobes or in the perihilar area and if there are also nodules in a perilymphatic distribution or if there is extensive mediastinal lymphadenopathy.
The presence of pleural plaques helps for the differentiation between IPF and asbestosis.
On the left a patient with UIP.
Notice the honeycombing and the preference of the subpleural and basal lung areas.
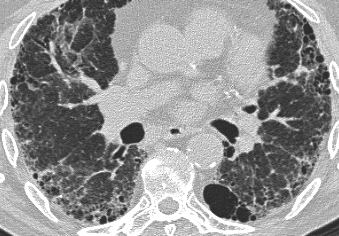 UIP: Typical case
UIP: Typical case
It is usually easy to recognize the pattern of UIP on HRCT.
NSIP
Nonspecific interstitial pneumonia (NSIP) is by some considered as a specific entity, with specific histologic characteristics, but by others as a 'wastebasket' diagnosis, representing cases of idiopathic interstitial pneumonia that cannot be classified as UIP, DIP, or OP.
NSIP is histologically characterized by a homogeneous, uniform pattern of cellular interstitial inflammation associated with variable degrees of fibrosis.
In contrast, UIP is associated with extensive fibrosis which is temporally inhomogeneous (i.e. various lesions are of different ages).
NSIP is a very inhomogeneous group.
NSIP ranges from type I which is a cellular pattern seen as ground glass opacity on HRCT to type IV with a fibrotic pattern, which may be indistinguishable from UIP.
On the left a patient with a NSIP.
This patient had a rash and muscle weakness.
Scroll through the images.
The predominant finding is ground glass opacity (GGO).
There is very subtle traction bronchiectasis, indicating that the GGO is the result of fibrosis and therefore irreversible.
It is important to note that we do not see the classic distribution of UIP, from which NSIP has to be differentiated.
The history of this patient is suggestive for the diagnosis dermatomyositis.
NSIP is by far the most common interstitial lung disease in patients with connective tissue disease.
NSIP (2)
NSIP is not a diagnosis on it's own.
It is a pattern of lung damage.
For the pathologist the key feature is the uniformity of the abnormality within the lung.
The role of the radiologist is more to 'exclude UIP pattern' rather than to make the diagnosis of NSIP.
The diagnosis of NSIP requires histological proof.
In all patients with a NSIP pattern, the clinician should be advised to look for connective tissue diseases, hypersensitivity pneumonitis or drugs .
On the left two cases of NSIP.
Note the varying combination of GGO and fibrosis (traction bronchiectasis), but the lack of honeycombing.
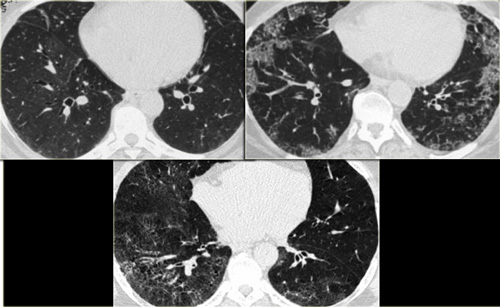
NSIP (3)
NSIP is the prevalent lung pattern in systemic sclerosis and polymyosisits/dermatomyositis (more than 90%), but also may occur in RA, SLE, Sj?gren's and MCTD.
In the images on your left you can appreciate again the spectrum of findings seen in NSIP.
All three patients were suffering from connective tissue disease, all cases were biopsy proven.
The first (top left) shows a very subtle GGO.
Note the difference in the density of the air within the bronchus and surrounding lungparenchyma (dark bronchus sign).
The second (top right) is a more obvious example of GGO with a superimposed fine reticular densities as a result of thickening of the intralobular septa.
The last image also shows GGO with a fine reticular pattern.
Notice the lack of honeycombing in all three cases, excluding UIP as diagnosis.
NSIP (4)
The HRCT of this patient with scleroderma and NSIP shows a fine subpleural reticular pattern in the upper lobes and more extensive abnormalities in the lower lung zones.
There are also areas of ground-glass and traction bronchiectases, but honeycombing is typically lacking.
Note also the mildly dilated esophagus, which is consistent with scleroderma.

COP
Cryptogenic organizing pneumonia (COP) used to be described as bronchiolitis obliterans with organizing pneumonia (BOOP) in an earlier version of the classification of idiopathic interstitial pneumonias.
It is a inflammatory process in which the healing process is characterized by organization of the exudate rather than by resorption ('unresolved pneumonia').
Organizing pneumonia is mostly idiopathic and then called cryptogenic, but is also seen in patients with pulmonary infection, drug reactions, collagen vascular disease, Wegener's granulomatosis and after toxic-fume inhalation.
OP presents with a several-month history of nonproductive cough, low-grade fever, malaise and shortness of breath.
There is a good response to corticosteroid therapy and a good prognosis.
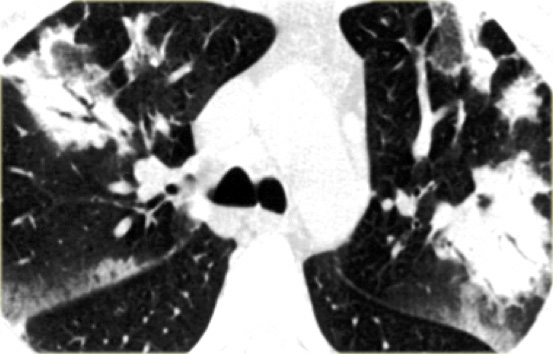
OP is again a great mimicker and can show a broad variety of HRCT findings, which makes it a frequent differential diagnosis and actually represents a diagnosis of exclusion.
Frequently biopsy is needed for final proof.
HRCT findings in OP
- Bilateral peripheral consolidations, sharply demarcated.
- Consolidations may be migratory.
- Lesions may show pleural tags or spiculae and give the impression of volume loss and slight retraction of the surrounding parenchyma (DD bronchogenic carcinoma).
- Bronchial wall thickening and dilatation are seen in most patients and are usually restricted to areas of consolidation or ground glass opacifications.
- Additional findings are pleural thickening, small pleural effusions and parenchymal bands.
On the left a patient with the typical bilateral peripheral consolidations of OP.
After exclusion of other diseases such as lymphoma, infection, bronchoalveolar carcinoma, the diagnosis of cryptogenic organizing pneumonia was made.
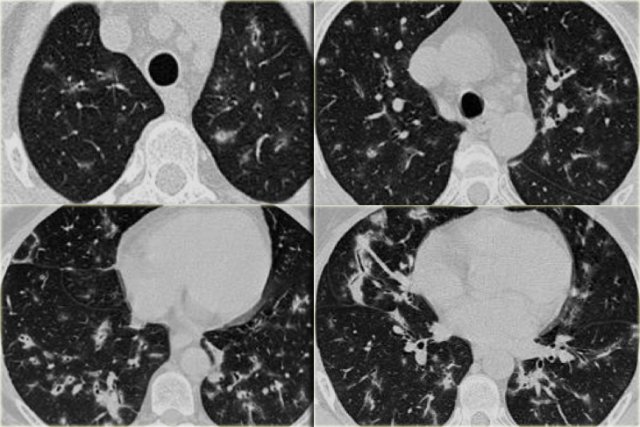 OP in a patient with collagen vascular disease
OP in a patient with collagen vascular disease
On the left a patient who complained of arthritic pain.
There are multiple small bilateral peripheral consolidations.
The findings in this patient are not as specific as in the former case, but this was also organizing pneumonia, but now related to collagen vascular disease.
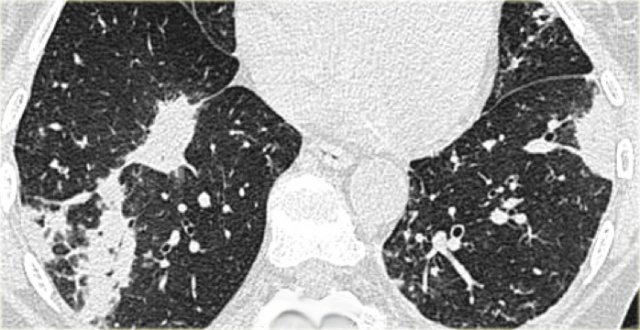 OP in rheumatoid arthritis
OP in rheumatoid arthritis
On the left a patient with rheumatoid arthritis and bilateral peripheral consolidations as a result of organizing pneumonia.
Patients with OP associated with collagen vascular diseases respond less well to therapy with steroids.
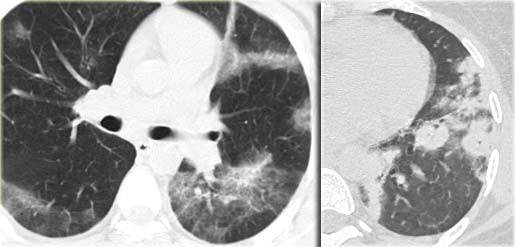 Chronic eosinophilic pneumonia (left) versus Organizing pneumonia (right)
Chronic eosinophilic pneumonia (left) versus Organizing pneumonia (right)
Differential diagnosis of Organizing Pneumonia.
- Chronic eosinophilic pneumonia
- Bronchoalveolar cell carcinoma
- Aspiration pneumonia
- Pulmonary infarction
- Lymphoma
The images on the left show the similarities between chronic eosinophilic pneumonia and organizing pneumonia.
Differentiation has to be made on the basis of clinical and laboratory findings.

RB-ILD and DIP
Respiratory bronchiolitis (RB), respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), and desquamative interstitial pneumonia (DIP) represent different degrees of severity of small airway and parenchymal reaction to cigarette smoke (8).
All smokers have various degrees of respiratory bronchiolitis, but it is usually asymptomatic.
However 5-10% of smokers have a clinically significant lung disease in association with RB, presenting with symptoms, lung function tests and auscultatory findings at clinical examination.
The term RB-ILD was proposed to describe the bronchocentric (or centrilobular) lung disease in these patients and the term DIP was used to describe the more diffuse disorder.
Radiologically however these diseases cannot be clearly separated because of the overlap of CT findings.
HRCT findings in RB-ILD
- Centrilobular nodules of ground glass opacity with upper lobe predominance
- Bronchial wall thickening
- Secondary lobuli with decreased attenuation (air trapping)
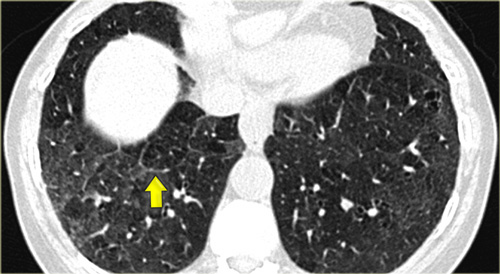
On the left a patient with RB-ILD.
The dominant feature is ground glass opacification and there are some thickened interlobular septa (arrow).
Usually these patients will also have smoking induced centrilobular emphysema and there is some evidence that respiratory bronchiolitis is the precursor of emphysema.
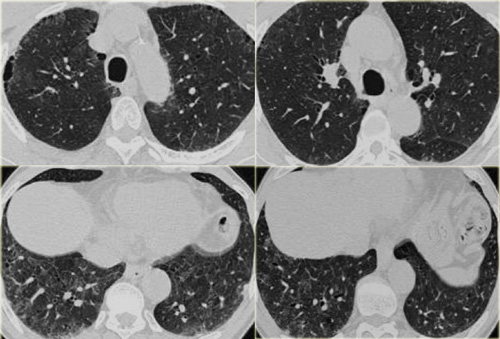
RB-ILD (2)
On the left a smoker with RB-ILD with subtle HRCT-findings.
The dominant pattern is ground glass opacification.
Additional findings in this patient are paraseptal emphysema in the upper lobes and some subtle septal thickening in the basal parts.
Based on these non-specific CT findings there is a broad differential diagnosis and additional clinical information is mandatory for the interpretion of the HRCT.
Since this patient is a smoker we first think RB-ILD.
In a immunocompromised patient PCP would be on top of the list.
If this patient was coughing up blood, this probably would be pulmonary hemorrhage (although we would expect more pulmonary densities in these patients).
If this patient was a bird-fancier we would first think hypersensitivity pneumonitis, but mostly these patients do not smoke.
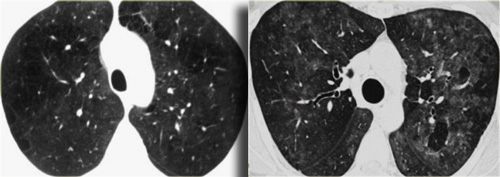 LEFT: RB-ILD in smoker. RIGHT: Hypersensitivity pneumonitis in non-smoker
LEFT: RB-ILD in smoker. RIGHT: Hypersensitivity pneumonitis in non-smoker
On the left two different patients with similarl HRCT findings.
The left one is a smoker with RB-ILD and the patient on the right has hypersensitivity pneumonits.
Note the difference in severity of ground glass opacities and the well defined areas of airtrapping in HP.
- If a patient is a smoker, think RB-ILD and look for additional smoking related features.
- If a patient is a non-smoker, think HP, and look at the expiratory CT scans.
Somehow smoking seems to protect against HP.
This is not a 100% specific criterium but is quite helpful for differential diagnosis.
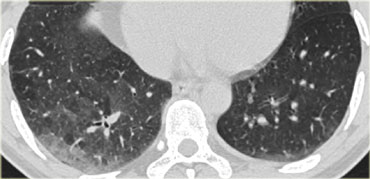
RB-ILD (3)
On the left a patient with DIP.
The HRCT shows diffuse areas of ground-glass density in the lower lobes and some mosaic pattern as the sole abnormality.
Reticular abnormalities and signs of fibrosis are typically absent.
These abnormalities are usually reversible and will disappear upon cessation of smoking.
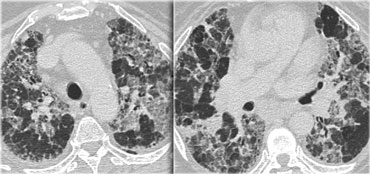 AIP
AIP
AIP
Acute interstitial pneumonia (AIP, earlier named Hamman Rich Pneumonitis) is a rare idiopathic lung disease characterized by diffuse alveolar damage with subsequent fibrosis.
It has a fatal outcome in many cases.
The histologic pattern aswell as the HRCT findings in AIP are indistinguishable from acute respiratory distress syndrome (ARDS).
The HRCT characteristics are diffuse or patchy consolidation, often with a crazy paving appearance like in the case on the left.
There are areas of consolidation and extensive areas of ground-glass density with a crazy-paving appearance.
These abnormalities developed in several days and this rapid progression of disease combined with these imaging findings are very suggestive of the diagnosis AIP.
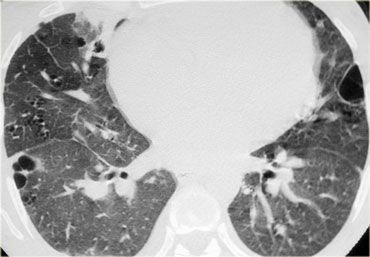
LIP
Lymphocytic interstitial pneumonitis or LIP is uncommon, being seen mainly in patients with autoimmune disease, particularly Sj?gren's syndrome, and in patients with AIDS.
Symptoms are nonspecific and often those of the patient's underlying disease
HRCT findings are usually nonspecific.
On the left a patient with Sjogren's syndrome with LIP.
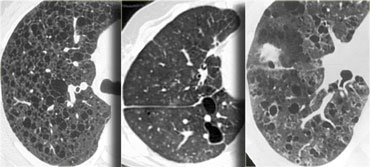 Lymphangiomyomatosis, LIP and Langerhans cell histiocytosis
Lymphangiomyomatosis, LIP and Langerhans cell histiocytosis
On the left three different patients with lung cysts.
From left to right: Lymphangiomyomatosis, LIP and Langerhans cell histiocytosis.
Drug-induced lung disease
Drug-induced lung disease is a major source of iatrogenic lung injury.
The major diagnostic problem is, that it may present with a large variety of radiologic patterns.
It may present as organizing pneumonia, eosinophilic pneumonia, fibrosis, hypersensitivity pneumonitis or even as ARDS.
The diagnosis of drug-induced pulmonary disease is usually one of exclusion.
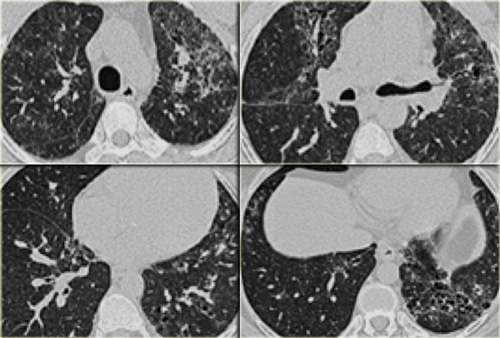 Drug-induced interstitial lung fibrosis
Drug-induced interstitial lung fibrosis
On the left a patient who is treated with cytotoxic drugs for a hematologic malignancy.
The radiographic findings are areas of ground glass opacity, some traction bronchiectasis and subtle honeycombing in the left lower lobe.
This could be the result of an idiopathic form of fibrosis like idiopathic pulmonary fibrosis and non-specific interstitial pneumonitis or fibrosis in chronic hypersensitivity pneumonitis and longstanding sarcoid.
However it is not one of the typical forms of fibrosis, that we commonly encounter in patients with a UIP pattern or NSIP pattern seen in collagenvascular diseases.
When there is fibrosis, that does not fit into any of the common diseases with fibrosis always consider drug-related lung disease in the differential.
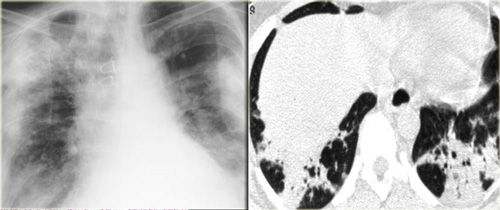 Drug-induced organizing pneumonia
Drug-induced organizing pneumonia
Drug-induced organizing pneumonia is commonly caused by Bleomycin and Cyclophosphamide and other drugs like Methotrexate, Amiodarone, Nitrofurantoin and Penicillamine (9).
The HRCT findings are the same as in cryptogenic organizing pneumonia.
Drug-induced non-specific interstitial pneumonita (NSIP) occurs most commonly as a manifestation of carmustine toxicity or of toxicity from noncytotoxic drugs such as amiodarone.
The radiologic findings are the same as in other forms of NSIP.
Uncommon interstitial lung diseases

Lymphangiomyomatosis
- Rare disease, that occurs only in premenopausal women
- Characterized by progressive proliferation of atypical muscle cells along the bronchioles leading to air trapping and the development of thin-walled cysts, that replace normal lung parenchyma.
- Identical pulmonary changes seen in 1% of patients with tuberous sclerosis (predominant involvement of young men).
Clinical findings:
- Majority of patients present with dyspnea.
- Chylous pleural effusions (40%), Pneumothorax (40%), hemoptysis (40%).
- Patients die within 10 years of the onset of symptoms.
- Pregnancy may exacerbate disease.
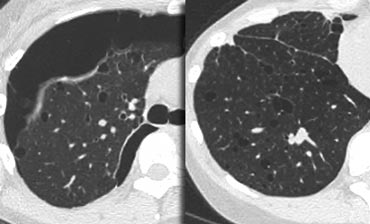
Key findings in Lymphangiomyomatosis:
- Numerous thin-walled cysts, surrounded by normal parenchyma.
- Cysts range from 2mm to 5cm in diameter, are round in shape and more or less uniform.
- Cysts are distributed diffusely throughout the lungs and upper and lower lobes are involved to a similar degree.
- Wall thickness of the cysts ranges from barely perceptible to 4 mm.
- Mediastinal or hilar adenopathy and pleural effusions (40%).
- Recurrent pneumothorax (40%).
On the left a typical case of LAM with multiple evenly spread thin walled cysts complicated by a pneumothorax.
 .
.
Differential diagnosis of Lymphangiomyomatosis:
- Langerhans cell histiocytosis: > 90% are smokers, cysts have irregular shapes and the basal costophrenic angles are spared.
- Centrilobular emphysema: characterized by airspaces that have no perceptible wall, centrilobular artery seen as dot in the centre.
- Lymphoid interstitial pneumonitis: seen in patients with HIV and Sj?gren syndrome.
On the left another typical case of LAM.

Langerhans cell histiocytosis
Langerhans cell histiocytosis is also known as pulmonary histiocytosis X or eosinophilic granuloma.
LCH is probably an allergic reaction to cigarette smoke since more than 90% of patients are active smokers.
In the early nodular stage it is characterized by a centrilobular granulomatous reaction by Langerhans histiocytes.
In the cystic stage bronchiolar obliteration causes alveolar wall fibrosis and cyst formation.
 Early stage Langerhans cell histiocytosis with small nodules
Early stage Langerhans cell histiocytosis with small nodules
HRCT findings in Langerhans cell histiocytosis:
- Early stage:
- Small irregular or stellate nodules in centrilobular location.
- Late stage (more commonly seen)
- Cystic airspaces Cysts have bizarre shapes, they may coalesce and than become larger.
- Upper and mid lobe predominance.
- Recurrent pneumothorax.
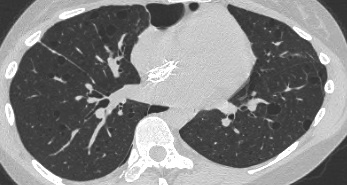
On the left early stage Langerhans cell histiocytosis with small nodules.
There are no cysts visible.
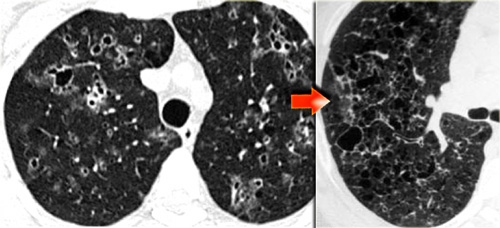 Late stage Langerhans' cell histiocytosis. Cysts progress to typical bizarre shaped cysts.
Late stage Langerhans' cell histiocytosis. Cysts progress to typical bizarre shaped cysts.
In a later stage the nodules start to cavitate and become cysts.
These cysts start as round structures but finally coalesce to become the typical bizarre shaped cysts of LCH.
In patients with LCH 95% have a smoking history.
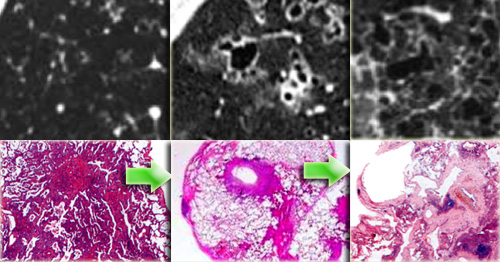 Specimen of Langerhans cell histiocytosis in three different stages
Specimen of Langerhans cell histiocytosis in three different stages
On the left radiological pathological correlation of Langerhans cell histiocytosis in respectively nodular stage and early and late cystic stage.
 Langerhans' cell histiocytosis
Langerhans' cell histiocytosis
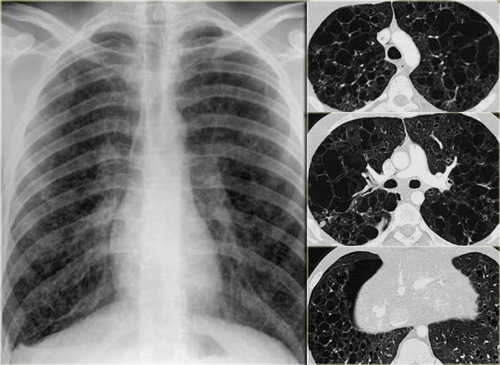
On the left a chest film of a 19 year old patient with Langerhans cell histiocytosis.
The dominant findig on the chest film is a reticular patern and that's about as far as you can go.
There is also hyperinflation.
No way you would have recognized that this pattern was caused by multiple cysts.
This is late stage Langerhans cell histiocytosis.
The most challenging differential diagnosis in this patient is centrilobular emphysema.
Emphysema however is defined as airspaces without definable walls.
Usually we can identify the central dot sign.
The upper lobe predominance is not helpfull in the differential as we can appreciate this in many inhalational diseases and also in emphysema.
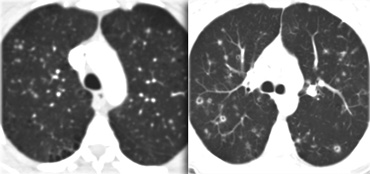 Langerhans cell histiosytosis: early phase and late phase
Langerhans cell histiosytosis: early phase and late phase
On the left another case of Langerhans' cell histiocytosis.
It started as small noduli, which progressed over time to cavitating nodules.
In the end this will progress to bizarre shaped cysts, that replace normal lung tissue.
Video
Images of a young male smoker with Langerhans cell histiocytosis.
Notice progression on second scan 7 years later.
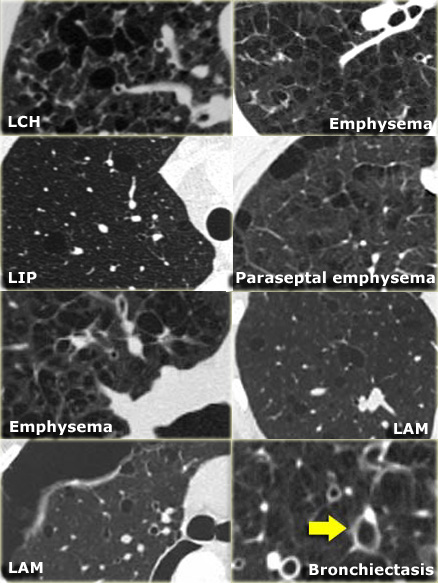
Differential diagnosis of Langerhans cell histiocytosis.
-
Nodular LCH:
- Sarcoidosis: perilymphatic distribution.
- Metastases: random distribution.
- Cystic LCH:
- LAM: round cysts, evenly distribution in women in the child-bearing age
- Cystic bronchiectasis: 'signet ring sign'.
- Centrilobular emphysema: no walls, central dot.
- LIP
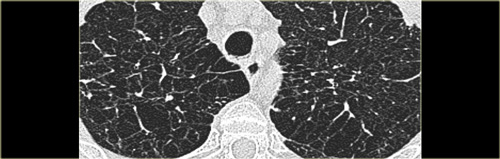 Emphysema mimicking Langerhans cell histiocytosis
Emphysema mimicking Langerhans cell histiocytosis
Emphysema, when it is severe, can mimick Langerhans cell histiosytosis.
When it extends beyond the centrilobular area to the edge of the secondary lobule, it may look as if it is cystic with walls.
In patients with LCH, the pathologist may find LCH, but also areas of emphysema, respiratory bronchiolitis and even fibrosis.
So these smoking-related diseases do not represent discrete entities.
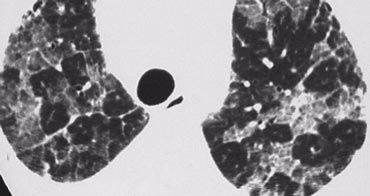 Alveolar proteinosis with crazy paving pattern
Alveolar proteinosis with crazy paving pattern
Alveolar proteinosis
Alveolar proteinosis is a rare disease characterized by filling of the alveolar spaces with PAS positive material due to an abnormality in surfactant metabolism.
The diagnosis is based on the suggestive HRCT pattern (crazy paving) and the characteristic features of BAL fluid (Broncho Alveolar Lavage)
- Usually between 30 and 50 years old.
- Nonproductive cough, fever, and mild dyspnea.
- Prognosis has improved since the advent of treatment using bronchoalveolar lavage.
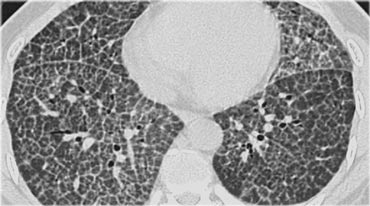
Key findings in alveolar proteinosis
- Crazy paving pattern: reticular pattern superimposed on ground glass opacification.
- Opacifications range from ground glass to consolidation.
On the left a typical case of alveolar proteinosis with extensive thickening of interlobular and intra-lobular septa.
This is caused by the fact that the proteinacious material, which is removed from the alveolar space by macrophages is transported to the interstitium and thus leads to thickening of septa.
The crazy paving pattern is a rather non-specific finding.
Many other diseases may present with this finding and are listed in the differential diagnosis.
Differential diagnosis of alveolar proteinosis
- Non cardiogenic edema: ARDS, Acute Interstitial Pneumonia.
-
Pneumonia:
- Infection (PCP and CMV).
- OP (organizing pneumonia).
- Chronic eosinophilic pneumonia.
- Hemorrhage.
- Bronchoalveolar ca.
Special Thanks
The authors like to thank Dr. Sujal Desai of the King's College Hospital in London for his inspiring lectures.
Some of the images used in this overview were provide by him.
We would also like to thank Dr. Richard Webb who produced such a fabulous educational CD (1).