HRCT - Basic Interpretation
Robin Smithuis, Otto van Delden and Cornelia Schaefer-Prokop
Radiology Department of the Rijnland Hospital, Leiderdorp and the Academical Medical Centre, Amsterdam, the Netherlands
Publicationdate
This article provides a practical approach to the interpretation of high-resolution computed tomography (HRCT) scans of the lungs.
The following topics will be addressed:
- Anatomy of the Secondary Pulmonary Lobule
- Basic HRCT Patterns
- Distribution of Abnormalities
- Differential Diagnosis of Interstitial Lung Diseases
Introduction
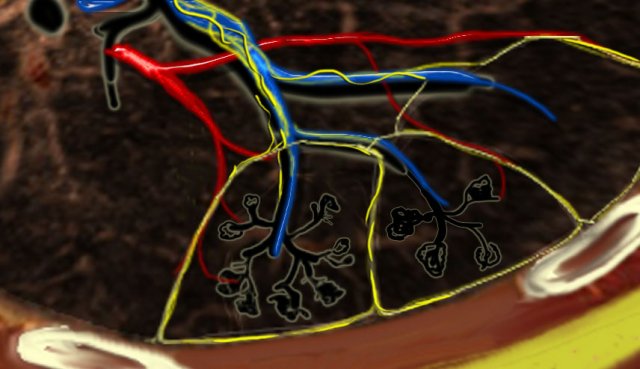 Secundary lobules. The centrilobular artery (in blue: oxygen-poor blood) and the terminal bronchiole run in the center. Lymphatics and veins (in red: oxygen-rich blood) run within the interlobular septa
Secundary lobules. The centrilobular artery (in blue: oxygen-poor blood) and the terminal bronchiole run in the center. Lymphatics and veins (in red: oxygen-rich blood) run within the interlobular septa
Anatomy of Secondary lobule
A thorough understanding of lung anatomy is essential for accurate HRCT interpretation. The diagnosis of interstitial lung diseases (ILDs) relies on identifying the specific involvement of the secondary pulmonary lobule, which is the fundamental anatomic and functional unit of the lung.
The secondary lobule is the smallest lung unit surrounded by connective tissue septa, measuring approximately 1–2 cm in diameter. It comprises 5–15 pulmonary acini, which contain the alveoli responsible for gas exchange. The lobule is supplied centrally by a small bronchiole or terminal bronchiole, accompanied by the centrilobular artery. The pulmonary veins and lymphatics run peripherally within the interlobular septa.
Under normal conditions, only a few of these thin septa are visible on HRCT. The lymphatic system of the lobule consists of two networks:
- A central network, which runs along the bronchovascular bundle toward the lobule’s center.
- A peripheral network, located within the interlobular septa and along the pleural linings.
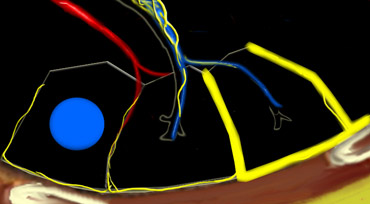 Centrilobular area in blue and perilymphatic area in yellow.
Centrilobular area in blue and perilymphatic area in yellow.
Key Anatomic Regions
- Centrilobular Area: The central portion of the secondary lobule, typically affected by diseases entering the lung via the airways (e.g., hypersensitivity pneumonitis, respiratory bronchiolitis, centrilobular emphysema).
- Perilymphatic Area: The peripheral portion of the secondary lobule, commonly involved in diseases affecting the lymphatics or interlobular septa (e.g., sarcoidosis, lymphangitic carcinomatosis, pulmonary edema). These diseases may also involve the central lymphatic network surrounding the bronchovascular bundle.

Basic HRCT Interpretation
A structured approach to HRCT interpretation involves addressing the following questions:
- What is the dominant HRCT pattern?
- Reticular (e.g., fibrosis)
- Nodular (e.g., granulomatous disease)
- High Attenuation (e.g., ground-glass opacities, consolidation)
- Low Attenuation (e.g., emphysema, cystic changes)
- Where is the abnormality located within the secondary lobule?
- Centrilobular
- Perilymphatic
- Random
- Is there a zonal predominance?
- Upper vs. lower lung zones
- Central vs. peripheral distribution
- Are there additional findings?
- Pleural effusion
- Lymphadenopathy
- Traction bronchiectasis
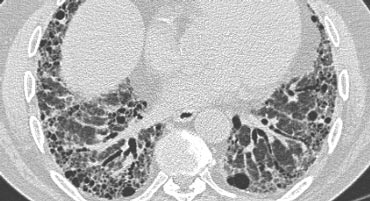 Typical UIP with honeycombing and traction bronchiectasis in a patient with idiopathic pulmonary fibrosis (IPF)
Typical UIP with honeycombing and traction bronchiectasis in a patient with idiopathic pulmonary fibrosis (IPF)
These morphologic findings must be correlated with the patient’s clinical history and physical examination.
It is important to recognize that HRCT is typically performed in a selected patient population—common conditions such as pneumonia, pulmonary embolism, cardiogenic edema, and lung carcinoma are often excluded beforehand.
Consequently, less common diseases—such as sarcoidosis, hypersensitivity pneumonitis, Langerhans cell histiocytosis, lymphangitic carcinomatosis, and usual interstitial pneumonia (UIP)—become more frequently encountered diagnoses on HRCT and may present as "Aunt Minnie" cases (classic, pathognomonic appearances).
Example
"Aunt Minnie" of typical UIP (Usual Interstitial Pneumonia)—a classic HRCT pattern characterized by subpleural, basilar-predominant reticular opacities with honeycombing and traction bronchiectasis.
Reticular pattern
In the reticular pattern, an excess of linear opacities is observed, resulting from either thickening of the interlobular septa or fibrotic changes, such as those seen in honeycombing.

Septal thickening
Thickening of the pulmonary interstitium—due to fluid accumulation, fibrous tissue deposition, or cellular infiltration—manifests as a reticular pattern on HRCT.
While septal thickening is relatively common in interstitial lung disease (ILD), it is rarely the predominant finding and has a limited differential diagnosis (see Table).
- Smooth Septal Thickening: Typically observed in:
- Interstitial pulmonary edema (e.g., Kerley B lines on chest radiography)
- Lymphangitic spread of carcinoma or lymphoma
- Alveolar proteinosis
- Nodular or Irregular Septal Thickening: Seen in:
- Lymphangitic spread of carcinoma or lymphoma
- Sarcoidosis
- Silicosis
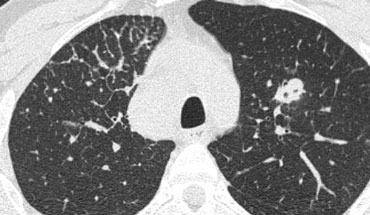 Focal septal thickening in lymphangitic carcinomatosis
Focal septal thickening in lymphangitic carcinomatosis
Case Example: Lymphangitic Carcinomatosis
Image
Focal, irregular septal thickening in the right upper lobe of a patient with known malignancy. This finding is classic for pulmonary lymphangitic carcinomatosis.
Supporting features include:
- Mediastinal lymphadenopathy
- A nodular lesion in the left lung, likely representing metastasis.
Pulmonary Lymphangitic Carcinomatosis (PLC):
- In 50% of cases, septal thickening is focal or unilateral, aiding differentiation from other causes (e.g., sarcoidosis, cardiogenic pulmonary edema).
- Hilar lymphadenopathy is visible in 50% of patients, and there is typically a history of malignancy.
- Similar findings may be seen in:
- Lymphoma
- Children with HIV infection developing lymphocytic interstitial pneumonitis (LIP), a rare benign lymphocytic infiltrative disease.
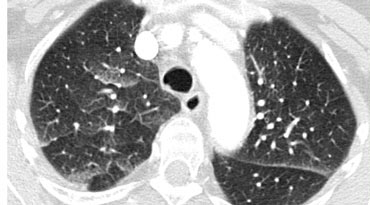 Septal thickening and ground-glass opacity with a gravitational distribution in a patient with cardiogenic pulmonary edema.
Septal thickening and ground-glass opacity with a gravitational distribution in a patient with cardiogenic pulmonary edema.
Case Example: Cardiogenic Pulmonary Edema
Image: A CT scan performed to rule out pulmonary embolism reveals a combination of smooth septal thickeningand ground-glass opacity with a gravitational distribution.
The diagnosis was cardiogenic pulmonary edema.
Key Features of Cardiogenic Pulmonary Edema:
- Typically presents with septal thickening + ground-glass opacity.
- Perihilar and gravitational distribution is characteristic of hydrostatic edema.
- Additional findings may include:
- Peribronchovascular interstitial thickening ("peribronchial cuffing")
- Fissural thickening
- Enlarged heart
- Pleural effusion
- While HRCT is not routinely used for diagnosis (clinical and radiographic findings usually suffice), unsuspected hydrostatic pulmonary edema may occasionally be identified.
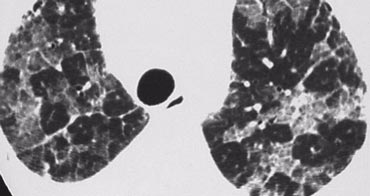 Alveolar proteinosis
Alveolar proteinosis
Case Example: Alveolar Proteinosis
Image: A patient with septal thickening + patchy ground-glass opacity. Some lobules are affected, while others are spared.
This combination is termed "crazy-paving".
Crazy-Paving Pattern:
- Initially considered pathognomonic for alveolar proteinosis, but now recognized in multiple conditions, including:
- Pneumocystis jirovecii pneumonia
- Bronchoalveolar carcinoma
- Sarcoidosis
- Nonspecific interstitial pneumonia (NSIP)
- Organizing pneumonia (COP)
- Adult respiratory distress syndrome (ARDS)
- Pulmonary hemorrhage
Alveolar Proteinosis:
- A rare diffuse lung disease of unknown etiology.
- Characterized by alveolar and interstitial accumulation of periodic acid-Schiff (PAS)-positive phospholipoproteinaceous material, derived from surfactant.
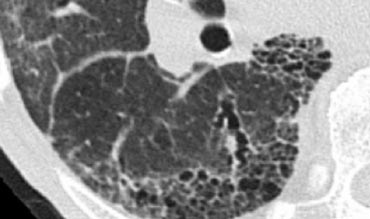 Honeycombing in a patient with UIP
Honeycombing in a patient with UIP
Honeycombing
Honeycombing constitutes the second recognizable reticular pattern on HRCT.
Due to its cystic appearance, honeycombing is also addressed in the context of low-attenuation patterns.
Pathologically, honeycombing is characterized by the presence of small cystic spaces lined by bronchiolar epithelium, with walls composed of dense fibrous tissue.
It is the hallmark feature of usual interstitial pneumonia (UIP).
Nodular pattern

Nodular Pattern Distribution on HRCT
The distribution of nodules observed on HRCT is the most critical factor in achieving an accurate diagnosis within the nodular pattern.
In most cases, small nodules can be categorized into one of three distributions: perilymphatic, centrilobular, or random. Random distribution refers to nodules that exhibit no preference for a specific location within the secondary pulmonary lobule.
The distribution of nodules shown on HRCT is the most important factor in making an accurate diagnosis in the nodular pattern:
- Perilymphatic distribution involves nodules located in relation to pleural surfaces, interlobular septa, and the peribronchovascular interstitium. These nodules are almost invariably visible in subpleural locations, particularly adjacent to the fissures.
- Centrilobular distribution is characterized by nodules confined to the centrilobular region. Unlike perilymphatic and random nodules, centrilobular nodules spare the pleural surfaces. The most peripheral nodules are typically centered 5–10 mm from fissures or the pleural surface.
- Random Distribution. Nodules in a random distribution are dispersed without preference relative to lung structures or the secondary lobule.
While they may involve pleural surfaces and fissures, they lack the subpleural predominance often observed in perilymphatic distributions.

Algorithm for nodular pattern
To distinguish between perilymphatic, random, and centrilobular nodules, the following algorithm is applied:
- Assess for the presence of pleural nodules, which are often most visible along the fissures.
- If pleural nodules are absent or minimal, the distribution is likely centrilobular.
- If pleural nodules are present, the pattern is either random (miliary) or perilymphatic.
- If pleural nodules are accompanied by nodules along the central bronchovascular interstitium and interlobular septa, the distribution is perilymphatic.
- If nodules are diffuse and uniformly distributed, the pattern is likely random.

Perilymphatic distribution
Perilymphatic nodules are most commonly associated with sarcoidosis.
They also occur in silicosis, coal worker’s pneumoconiosis, and lymphangitic carcinomatosis.
There is an overlap in the differential diagnosis of perilymphatic nodules and nodular septal thickening within the reticular pattern, sometimes referred to as a reticulonodular pattern.
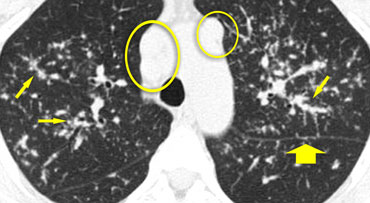
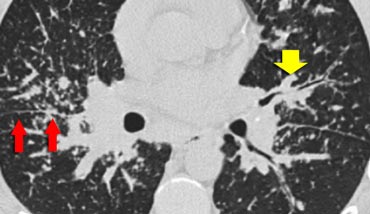 Sarcoidosis
Sarcoidosis
Case 1
The image shows a typical case of perilymphatic nodule distribution in a patient with sarcoidosis.
Note the nodules along the fissures (red arrows), indicative of a perilymphatic distribution.
Careful examination for these nodules in the subpleural region and along the fissures is essential, as this finding is highly specific for sarcoidosis.
Typically, sarcoidosis demonstrates an upper lobe and perihilar predominance, with the majority of nodules located along the bronchovascular bundle (yellow arrow).
 Sarcoidosis
Sarcoidosis
Case 2
The image shows another typical case of sarcoidosis.
Perilymphatic nodules (arrows) are accompanied by multiple enlarged lymph nodes (circles)., a characteristic feature of the disease.
In end-stage sarcoidosis, fibrosis predominantly affects the upper lobes and perihilar regions.
 Ill defined centrilobular nodules of ground glass density in a patient with hypersensitivity pneumonitis
Ill defined centrilobular nodules of ground glass density in a patient with hypersensitivity pneumonitis
Centrilobular distribution
Centrilobular nodules are observed in diseases that enter the lung via the airways.
Pathogens access the central area of the secondary lobule through the terminal bronchiole, leading to conditions such as:
- Hypersensitivity pneumonitis
- Respiratory bronchiolitis in smokers
- Infectious airway diseases (e.g., endobronchial spread of tuberculosis or nontuberculous mycobacteria, bronchopneumonia)
- Less commonly in bronchioloalveolar carcinoma, pulmonary edema, and vasculitis.
In many cases, these nodules present as ill-defined, ground-glass density lesions (figure) and are sometimes referred to as acinar nodules.

Tree-in-Bud Pattern
In the context of centrilobular nodules, the identification of the "tree-in-bud" pattern is valuable for narrowing the differential diagnosis.
The tree-in-bud pattern describes an irregular, often nodular branching structure, most readily visualized in the lung periphery.
Pathologically, it represents dilated and impacted (mucus- or pus-filled) centrilobular bronchioles.

The presence of a tree-in-bud pattern almost invariably indicates:
- Endobronchial spread of infection, such as tuberculosis (TB), Mycobacterium avium complex (MAC), or bacterial bronchopneumonia.
- Airway diseases associated with infection, including cystic fibrosis and bronchiectasis.
- Less commonly, airway diseases primarily associated with mucus retention, such as allergic bronchopulmonary aspergillosis (ABPA) and asthma.
- Aspiration.
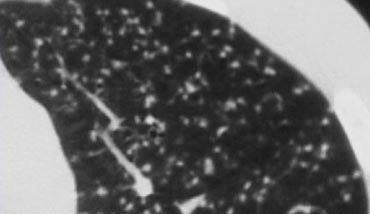
 Typical Tree-in-bud appearance in a patient with active TB.
Typical Tree-in-bud appearance in a patient with active TB.
Example
In the image on the left, a tree-in-bud pattern is demonstrated.
In the appropriate clinical context, this finding should raise suspicion for active endobronchial spread of TB.
Notably, in most patients with active tuberculosis, HRCT often reveals evidence of bronchogenic dissemination of disease even before bacteriologic confirmation is available (6).
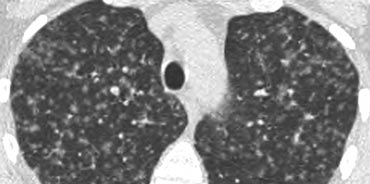
 Random distribution of nodules in miliary tuberculosis
Random distribution of nodules in miliary tuberculosis
Random distribution
Small, randomly distributed nodules are observed in:
- Hematogenous metastases
- Miliary tuberculosis
- Miliary fungal infections
- Sarcoidosis
- Langerhans cell histiocytosis (LCH) (early nodular stage)
While sarcoidosis typically exhibits a perilymphatic distribution, extensive disease can spread along the lymphatics within the bronchovascular bundle to the lung periphery, potentially reaching the centrilobular area.
This may result in a combined perilymphatic-centrilobular pattern, which can mimic a random distribution.
Example
The image on the left depicts a patient with randomly distributed nodules secondary to miliary TB.
The random distribution results from hematogenous dissemination of the infection.
 Langerhans cell histiocytosis: early nodular stage before the typical cysts appear.
Langerhans cell histiocytosis: early nodular stage before the typical cysts appear.
Example
The image presented demonstrates a typical random nodular pattern in a patient with Langerhans cell histiocytosis (LCH).
LCH is a rare disease characterized by multiple irregular cysts, predominantly in patients with a history of nicotine abuse.
In its early phase, LCH manifests as a nodular disease (figure).
Over time, these nodules cavitate and evolve into cysts.
As with other smoking-related diseases, there is a predominance in the upper lobes.
High Attenuation pattern
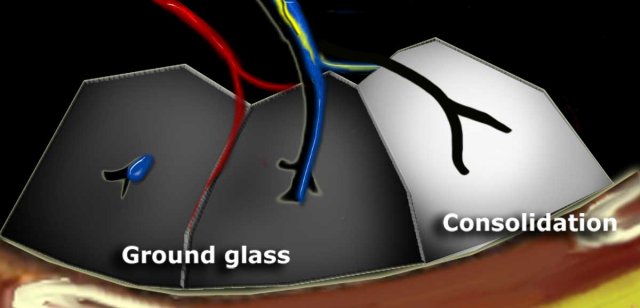 Dark bronchus sign in ground glass opacity. Complete obscuration of vessels in consolidation and air bronchogram.
Dark bronchus sign in ground glass opacity. Complete obscuration of vessels in consolidation and air bronchogram.
Increased lung attenuation is classified as ground-glass opacity (GGO) when there is a hazy increase in lung opacity that does not obscure the underlying vessels. If the increased opacity obscures the vessels, it is termed consolidation. In both GGO and consolidation, the increased lung density results from the replacement of alveolar air by fluid, cells, or fibrosis.
Dark Bronchus Sign
In GGO, air-filled bronchi appear abnormally dark against the surrounding, abnormally opaque lung tissue, which still contains some air. This is known as the dark bronchus sign, visible only on high-resolution computed tomography (HRCT), and it highlights underlying lung pathology.
Air Bronchogram
Conceptually similar to the dark bronchus sign, an air bronchogram occurs in complete consolidation, where the surrounding lung tissue is significantly denser. This sign is visible on both HRCT and chest X-rays.

Ground-glass opacity
Ground-glass opacity (GGO) represents:
- Filling of alveolar spaces with pus, edema, hemorrhage, inflammatory cells, or tumor cells.
- Thickening of the interstitium or alveolar walls below the spatial resolution of HRCT, as seen in fibrosis.
Thus, GGO may result from airspace disease (alveolar filling) or interstitial lung disease (e.g., fibrosis).
The location of abnormalities in GGO can provide diagnostic clues:
- Upper zone predominance: Respiratory bronchiolitis, Pneumocystis pneumonia.
- Lower zone predominance: Usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia (NSIP), desquamative interstitial pneumonia (DIP).
- Centrilobular distribution: Hypersensitivity pneumonitis, respiratory bronchiolitis.
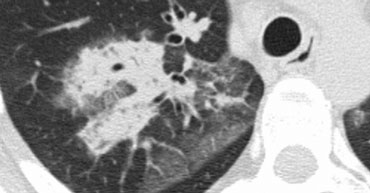 Broncho-alveolar cell carcinoma with ground-glass opacity and consolidation
Broncho-alveolar cell carcinoma with ground-glass opacity and consolidation
The GGO pattern itself is relatively nonspecific. Unsurprisingly, there is significant overlap in the causes of GGO and consolidation, and some diseases may present with both GGO and consolidation.
Example
In the image on the left, consolidation and GGO are observed in a patient with persistent chest abnormalities and weight loss, without signs of infection.
This presentation suggested a chronic disease.
The absence of honeycombing or traction bronchiectasis ruled out fibrosis, while the weight loss raised suspicion for malignancy.
Histology confirmed bronchioloalveolar cell carcinoma (BAC).
BAC may present as:
- A solitary nodule or mass (40% of patients).
- Focal or diffuse consolidation (30%), as in this case.
- Diffuse, ill-defined centrilobular nodules (30%) due to endobronchial spread.
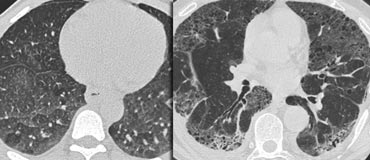 LEFT: No fibrosis, so potentially treatable lung disease. RIGHT: Fibrosis, so no treatable lung disease.
LEFT: No fibrosis, so potentially treatable lung disease. RIGHT: Fibrosis, so no treatable lung disease.
Treatable or not treatable?
While GGO is nonspecific, it is a highly significant finding: 60–80% of patients with GGO on HRCT have an active and potentially treatable lung disease.
In the remaining 20–40% of cases, the GGO reflects fibrosis, and the disease is not treatable.
In these cases, associated HRCT findings of fibrosis, such as traction bronchiectasis and honeycombing, are typically present.
Example
The images show two cases with GGO: one without fibrosis (potentially treatable) and another with traction bronchiectasis (indicating fibrosis).
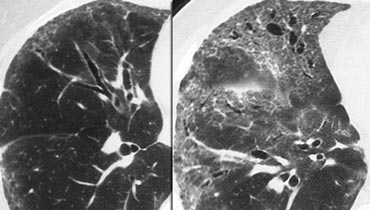 Non specific interstitial pneumonitis (NSIP). Notice lower lobe predominance.
Non specific interstitial pneumonitis (NSIP). Notice lower lobe predominance.
Example
The images depict a patient with GGO as the dominant pattern, accompanied by traction bronchiectasis, indicating fibrosis.
This presentation is consistent with one of the possible patterns of nonspecific interstitial pneumonia (NSIP).
NSIP is histologically characterized by a relatively uniform pattern of cellular interstitial inflammation with variable degrees of fibrosis.
Like UIP, NSIP primarily affects the dependent regions of the lower lobes but lacks the extensive fibrosis and honeycombing seen in UIP.
NSIP may be idiopathic or associated with collagen vascular diseases or exposure to drugs or chemicals.
It carries a relatively good prognosis, with the majority of patients responding to treatment with corticosteroids—unlike UIP, which has a poor prognosis.

Mosaic attenuation
The term mosaic attenuation describes density differences between affected and unaffected lung regions, resulting in a patchy appearance of black and white lung areas. The radiologist’s role is to determine which component—the black or the white lung—is abnormal.
When ground-glass opacity (GGO) presents as mosaic attenuation, consider the following possibilities:
- Infiltrative processes adjacent to normal lung.
- Normal lung appearing relatively dense adjacent to lung regions with air trapping.
- Hyperperfused lung adjacent to hypoperfused lung due to chronic thromboembolic disease.
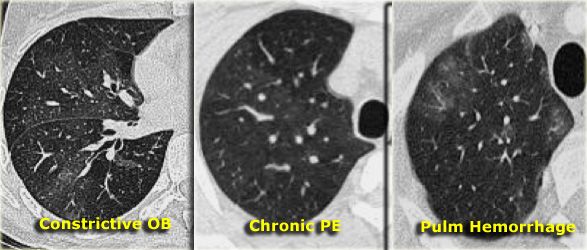 Three different causes of mosaic attenuation
Three different causes of mosaic attenuation
Differentiating these entities can be challenging. Two diagnostic clues aid further distinction:
- Expiratory scans for air trapping.
- Vessel assessment:
- If vessels are less visible in the "black" lung compared to the "white" lung, the "black" lung is likely abnormal. This suggests either obstructive bronchiolitis or chronic pulmonary embolism. An expiratory scan may help differentiate these conditions.
- If vessels appear similar in both "black" and "white" lung regions, the patient likely has infiltrative lung disease, such as pulmonary hemorrhage (as shown on the right).
Temporary bronchiolitis with air trapping is observed in:
- (Post-)infectious states
- Inhalation of toxins
- Rheumatoid arthritis, Sjögren syndrome
- Post-transplant settings
- Drug reactions (e.g., penicillamine)
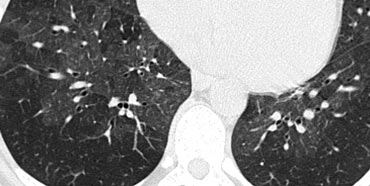 Mosaic pattern in a patient with hypersensitivity pneumonitis
Mosaic pattern in a patient with hypersensitivity pneumonitis
Mosaic Attenuation in Hypersensitivity Pneumonitis
The image on the left shows a patient with a ground-glass pattern in a mosaic distribution. Some lobules are involved, while others are spared.
The differential diagnosis includes hypersensitivity pneumonitis (HP), bronchiolitis, or thromboembolic disease.
The clinical history was typical for HP.
HP usually presents with centrilobular nodules of ground-glass density (acinar nodules).
When confluent, HRCT demonstrates diffuse ground-glass opacity.
HP is an allergic lung disease caused by the inhalation of antigens present in various organic dusts.
Farmer’s lung, the most well-known HP syndrome, results from inhaling fungal organisms in moist hay or exposure to pet birds.
HP typically manifests in two forms:
- Ground-glass opacity in a mosaic distribution (as in this case).
- Centrilobular nodules of ground-glass density (acinar nodules).
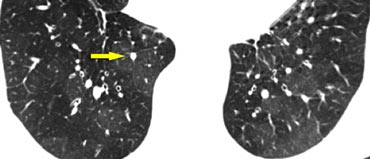 Mosaic pattern in a patient with chronic thromboemboli
Mosaic pattern in a patient with chronic thromboemboli
Mosaic Attenuation in Chronic Thromboembolic Disease
The image on the left depicts a patient with a ground-glass pattern in a mosaic distribution.
The key finding here is the enlargement of pulmonary arteries (arrow) in areas of ground-glass opacity.
This appearance results from hyperperfused lung adjacent to oligemic lung with reduced vessel caliber due to chronic thromboembolic disease.

Another patient (left) demonstrates a similar ground-glass pattern in a mosaic distribution.
The ground-glass appearance is due to hyperperfused lung with large vessels adjacent to oligemic lung (yellow arrow) with small vessels caused by chronic thromboembolic disease.
Mural emboli and intravascular septa are typical features of chronic thromboemboli, reflecting partial recanalization (blue arrow).
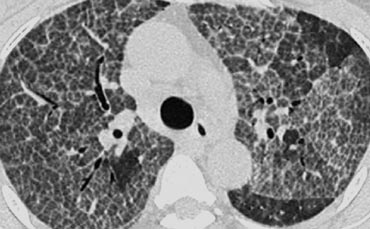 Crazy Pavin in a patient with Alveolar proteinosis.
Crazy Pavin in a patient with Alveolar proteinosis.
Crazy Paving
The crazy paving pattern combines ground-glass opacity with superimposed septal thickening.
Initially thought to be specific for alveolar proteinosis, it has since been observed in other conditions, including:
- Alveolar proteinosis
- Sarcoidosis
- Nonspecific interstitial pneumonia (NSIP)
- Organizing pneumonia (COP/BOOP)
- Infections (e.g., Pneumocystis jirovecii pneumonia, viral, Mycoplasma, bacterial)
- Neoplasms (e.g., bronchioloalveolar carcinoma)
- Pulmonary hemorrhage
- Edema (e.g., heart failure, ARDS, acute interstitial pneumonia)

Consolidation
Consolidation is synonymous with airspace disease.
When considering the causes of consolidation, the key question is: "What is replacing the air in the alveoli?"
Possible replacements include pus, edema, blood, or tumor cells. Even fibrosis, as seen in UIP, NSIP, and long-standing sarcoidosis, can replace alveolar air and result in consolidation.
Acute consolidation is seen in:
- Pneumonias (bacterial, mycoplasma, PCP)
- Pulmonary edema due to heart failure or ARDS
- Hemorrhage
- Acute eosinophilic pneumonia
Chronic consolidation is seen in:
- Organizing Pneumonia
- Chronic eosinophilic pneumonia
- Fibrosis in UIP and NSIP
- Bronchoalveolar carcinoma or lymphoma
Most patients who are evaluated with HRCT, will have chronic consolidation, which limits the differential diagnosis.
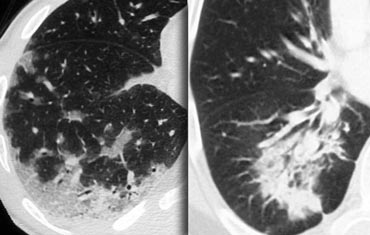 Two patients with chronic consolidations as a result of COP (cryptogenic organizing pneumonia)
Two patients with chronic consolidations as a result of COP (cryptogenic organizing pneumonia)
On the left two cases with chronic consolidation.
There are patchy non-segmental consolidations in a subpleural and peripheral distribution.
The differential diagnosis is the same as the list above.
The final diagnosis was cryptogenic organizing pneumonia (COP).
In chronic eosinophilic pneumonia the HRCT findings will be the same, but there will be eosinophilia.
In fibrosis there will be other signs of fibrosis like honeycombing or traction bronchiectasis.
Bronchoalveolar carcinoma can also look like this.
Organizing pneumonia (OP)
Organizing pneumonia represents an inflammatory process in which the healing process is characterized by organization and cicatrization of the exudate rather than by resolution and resorption.
It is also described as 'unresolved pneumonia'.
If no cause can be identified it is called cryptogenic organizing pneumonia (COP).
It was described in earlier years as Bronchiolitis-obliterans-organizing pneumonia (BOOP).
Patients with COP typically present with a several-month history of nonproductive cough.
Many cases are idiopathic, but OP may also be seen in patients with pulmonary infection, drug reactions, collagen vascular disease, Wegener's granulomatosis and after toxic-fume inhalation.
 Chronic eosinophilic granuloma
Chronic eosinophilic granuloma
On the left a case of chronic eosinophilic pneumonia.
It was a patient with low-grade fever, progressive shortness of breath and an abnormal chest radiograph.
There was a marked eosinophilia in the peripheral blood.
Like in COP we see patchy non-segmental consolidations in a subpleural distribution.
Chronic eosinophilic pneumonia is an idiopathic condition characterized by extensive filling of alveoli by an infiltrate consisting primarily of eosinophils.
Chronic eosinophilic pneumonia is usually associated with an increased number of eosinophils in the peripheral blood and patients respond promptly to treatment with steroids.
Low Attenuation pattern

The fourth pattern includes abnormalities that result in decreased lung attenuation or air-filled lesions.
These include:
- Emphysema
- Lung cysts (LAM, LIP, Langerhans cell histiocytosis)
- Bronchiectasis
- Honeycombing
Most diseases with a low attenuation pattern can be readily distinguished on the basis of HRCT findings.
 Centrilobular emphysema due to smoking. The periphery of the lung is spared (blue arrows). Centrilobular artery (yellow arrows) is seen in the center of the hypodense area.
Centrilobular emphysema due to smoking. The periphery of the lung is spared (blue arrows). Centrilobular artery (yellow arrows) is seen in the center of the hypodense area.
Emphysema
Emphysema typically presents as areas of low attenuation without visible walls as a result of parenchymal destruction.
-
Centrilobular emphysema
- Most common type
- Irreversible destruction of alveolar walls in the centrilobular portion of the lobule
- Upper lobe predominance and uneven distribution
- Strongly associated with smoking.
-
Panlobular emphysema
- Affects the whole secondary lobule
- Lower lobe predominance
- In alpha-1-antitrypsin deficiency, but also seen in smokers with advanced emphysema
-
Paraseptal emphysema
- Adjacent to the pleura and interlobar fissures
- Can be isolated phenomenon in young adults, or in older patients with centrilobular emphysema
- In young adults often associated with spontaneous pneumothorax
 Paraseptal emphysema with small bullae
Paraseptal emphysema with small bullae
Paraseptal emphysema
Paraseptal emphysema is localized near fissures and pleura and is frequently associated with bullae formation (area of emphysema larger than 1 cm in diameter).
Apical bullae may lead to spontaneous pneumothorax.
Giant bullae occasionally cause severe compression of adjacent lung tissue.
 Panlobular emphysema
Panlobular emphysema
Panlobular emphysema
On the left a typical case of panlobular emphysema.
There is uniform destruction of the underlying architecture of the secondary pulmonary lobules, leading to widespread areas of abnormally low attenuation.
Pulmonary vessels in the affected lung appear fewer and smaller than normal.
Panlobular emphysema is diffuse and is most severe in the lower lobes.
In severe panlobular emphysema, the characteristic appearance of extensive lung destruction and the associated paucity of vascular markings are easily distinguishable from normal lung parenchyma.
On the other hand, mild and even moderately severe panlobular emphysema can be very subtle and difficult to detect on HRCT(1).

Cystic lung disease
Lung cysts are defined as radiolucent areas with a wall thickness of less than 4mm.
Cystic lung diseases as listed in the table on the left.
Cavities are defined as radiolucent areas with a wall thickness of more than 4mm and are seen in infection (TB, Staph, fungal, hydatid), septic emboli, squamous cell carcinoma and Wegener's disease.
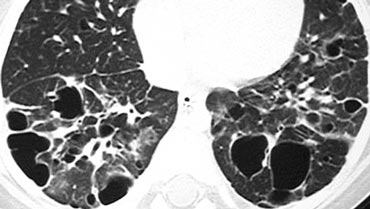 Langerhans cell histiocytosis
Langerhans cell histiocytosis
On the left a case with multiple round and bizarre shaped cysts.
There was an upper lobe predominance.
The patient had a long history of smoking.
This combination of findings is typical for Langerhans cell histiocytosis.
Langerhans cell histiocytosis (LCH) is an idiopathic disease characterized in its early stages by granulomatous nodules containing Langerhans histiocytes and eosinophils.
In its later stages, the granulomas are replaced by fibrosis and the formation of cysts.
It is an uncommon condition.
The majority of patients are young or middle-aged adults presenting with nonspecific symptoms of cough and dyspnea. Up to 20% of patients present with pneumothorax and over 90% of patients are smokers.
Most cysts appear round, but can also have bizarre shapes (bilobed or clover-leaf shaped).
An upper lobe predominance in the size and number of cysts is common.
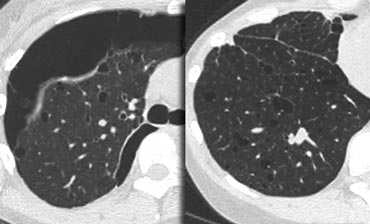 Lymphangiomyomatosis complicated by pneumothorax
Lymphangiomyomatosis complicated by pneumothorax
On the left a case with multiple cysts that are evenly distributed througout the lung ( in contrast to LCH).
Notice the pneumothorax.
There was no history of smoking and this was a 40 year old female.
This combination of findings is typical for Lymphangiomyomatosis (LAM).
Lymphangiomyomatosis is a rare disease characterized by progressive proliferation of spindle cells, resembling smooth muscle.
Proliferation of these cells along the bronchioles leads to air trapping and the development of thin-walled lung cysts.
Rupture of these cysts can result in pneumothorax.
Other features of LAM include adenopathy and pleural effusion.
Lymphangiomyomatosis occurs only in women, usually of child-bearing age, between 17 and 50 years.
Identical clinical, radiologic, and pathologic pulmonary changes are seen in about 1% of patients with tuberous sclerosis.
Most patients die within 10 years of the onset of symptoms.

Bronchiectasis
Bronchiectasis is defined as localized bronchial dilatation.
The diagnosis of bronchiectasis is usually based on a combination of the following findings:
- bronchial dilatation (signet-ring sign)
- bronchial wall thickening
- lack of normal tapering with visibility of airways in the peripheral lung
- mucus retention in the broncial lumen
- associated atelectasis and sometimes air trapping
A signet-ring sign represents an axial cut of a dilated bronchus (ring) with its accompanying small artery (signet).
The most common cause of bronchiectasis is prior infection, usually viral, at an early age.
It also occurs in patients with chronic bronchitis, COPD and cystic fibrosis.
Bronchiectasis may mimic cystic lung disease and bullous emphysema.
Bronchiectasis caused by primary airway disease should be differentiated from tracion bronchiectasis as a result of fibrosis.
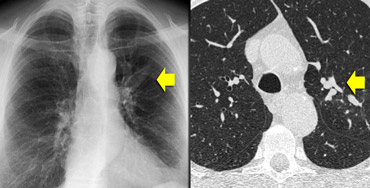 ABPA: glove-finger shadow due to mucoid impaction in central bronchiectasis in a patient with asthma.
ABPA: glove-finger shadow due to mucoid impaction in central bronchiectasis in a patient with asthma.
On the left we see a chest film with a typical finger-in-glove shadow.
The HRCT shows focal bronchiectasis with extensive mucoid impaction, which is in the appropriate clinical setting (asthma and serum eosinophilia) typical for Allergic bronchopulmonary aspergillosis (ABPA).
Allergic bronchopulmonary aspergillosis is a lung disease occurring in patients with asthma or cystic fibrosis, triggered by a hypersensitivity reaction to the presence of Aspergillus fumigatus in the airways.
It characteristically presents with the findings of central bronchiectasis, mucoid impaction and atelectasis.

Honeycombing
Honeycombing is defined by the presence of small cystic spaces with irregularly thickened walls composed of fibrous tissue.
Honeycomb cysts often predominate in the peripheral and subpleural lung regions regardless of their cause.
Subpleural honeycomb cysts typically occur in several contiguous layers.
This finding can allow honeycombing to be distinguished from paraseptal emphysema in which subpleural cysts usually occur in a single layer.
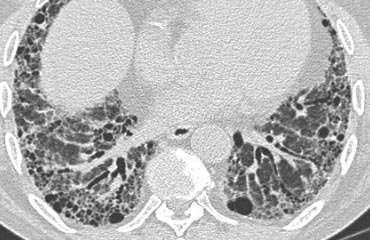 Honeycombing and traction bronchiectasis in UIP.
Honeycombing and traction bronchiectasis in UIP.
The case on the left shows subpleural honeycomb cysts in several contiguous layers.
There is also a lower lobe predominance and widespread traction bronchiectasis.
These findings are typical for Usual Interstitial Pneumonia (UIP).
UIP or 'end-stage lung' is a pathology diagnosis and usually shown at lungbiopsy, when honeycombing is visible.
Idiopathic pulmonary fibrosis (IPF), accounts for more than 60% of the cases of UIP.
UIP with lung fibrosis is also a common pattern of auto-immune disease and drug-related lung injury.
A long list of drugs have been implicated, but this pattern is most commonly the result of cytotoxic chemotherapeutic agents such as bleomycin, busulfan, vincristine, methotrexate, adriamycin, and carmustine (BCNU).
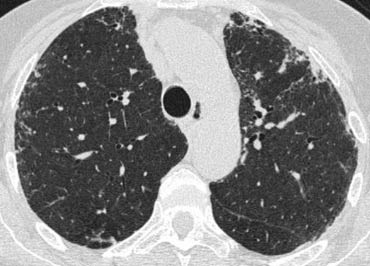
On the left another case of UIP.
The lower zone predominance is demonstrated when you scroll through the images.
Notice the ground glass opacity in the left lower lobe as a result of fibrous tissue replacing the air in the alveoli.
Distribution within the lung

Upper versus lower zone distribution
Upper lung zone preference is mostly seen in inhalation diseases:
- Inhaled particles: pneumoconiosis (silica or coal)
- Smoking related diseases (centrilobular emphysema)
- Respiratory bronchiolitis (RB-ILD)
- Langerhans cell histiocytosis
- Hypersensitivity pneumonitis
- Sarcoidosis
Lower zone preference is seen in:
- UIP
- Aspiration
- Pulmonary edema

Central versus peripheral distribution
Central distribution is seen in sarcoidosis, bronchitis and cardiogenic pulmonary edema.
Peripheral distribution is mainly seen in cryptogenic organizing pneumonia (COP), chronic eosinophilic pneumonia and UIP.
Additional findings

Pleural effusion
Diseases that manifest with pleural effusion are listed in the table.

Lymphadenopathy
In sarcoidosis the common pattern is right paratracheal and bilateral hilar adenopathy ('1-2-3-sign').
In lung carcinoma and lymphangitic carcinomatosis adenopathy is usually unilateral.
Eggshell calcification
This is commonly seen in lymph nodes in patients with silicosis and coal-worker's pneumoconiosis and is sometimes seen in sarcoidosis, postirradiation Hodgkin disease, blastomycosis and scleroderma.
Differential diagnosis of interstitial lung diseases

Examples of reticular pattern:
- Lymphangitic carcinomatosis: irregular septal thickening, usually focal or unilateral 50% adenopathy', known carcinoma.
- Cardiogenic pulmonary edema: incidental finding in HRCT, smooth septal thickening with basal predominance (Kerley B lines), ground-glass opacity with a gravitational and perihilar distribution, thickening of the peribronchovascular interstitium (peribronchial cuffing)
- Lymphangitic carcinomatosis
- Lymphangitic carcinomatosis with hilar adenopathy.
- Alveolar proteinosis: ground glass attenuation with septal thickening (crazy paving).
- Cardiogenic pulmonary edema.

Examples of nodular pattern
- Hypersensitivity pneumonitis: ill defined centrilobular nodules.
- Miliary TB: random nodules
- Sarcoidosis: nodules with perilymphatic distribution, along fissures, adenopathy.
- Hypersensitivity pneumonitis: centrilobular nodules, notice sparing of the area next to pleura and fissure.

More nodular pattern
- Sarcoidosis: nodules with perilymphatic distribution, along fissures, adenopathy.
- TB: Tree-in-bud appearance in a patient with active TB.
- Langerhans cell histiocytosis: early nodular stage before the typical cysts appear.
- Respiratory bronchiolitis in infection.

Examples of High Attenuation pattern
- Chronic eosinophilic pneumonia with peripheral areas of ground glass opacity.
- Sarcoid end-stage with massive fibrosis in upper lobes presenting as areas of consolidation. Notice lymphadenopathy.
- Chronic eosinophilic pneumonia with peripheral areas of consolidation.
- Broncho-alveolar cell carcinoma with both areas of ground glass opacity and consolidation

High Attenuation pattern (2)
- Non specific interstitial pneumonitis (NSIP): ground glass with traction bronchiectasis, no honeycombing.
- Cryptogenic organizing pneumonia (COP).
- Sarcoidosis end-stage: consolidation as a result of massive fibrosis perihilar and in upper lobes.
- COP.

Low Attenuation pattern
- Lymphangiomyomatosis (LAM): uniform cysts in woman of child-bearing age; no history of smoking; adenopathy and pleural effusion; sometimes pneumothorax.
- LCH: multiple round and bizarre shaped cysts; smoking history.
- Honeycombing
- Centrilobular emphysema: low attenuation areas without walls.

Low Attenuation pattern (2)
- Centrilobular emphysema: low attenuation areas without walls. Notice the centrilobular artery in the center.
- Langerhans cell histiocytosis (LCH): multiple thick walled cysts; smoking history.
- Honeycombing.
- Lymphangiomyomatosis (LAM): regular cysts in woman of child-bearing age.